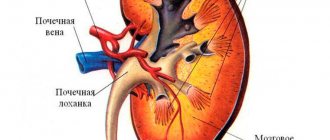
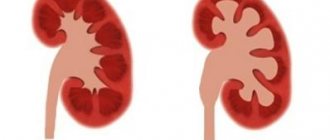
Polycystic kidney disease is a genetic pathology characterized by the degeneration of the renal parenchyma (functional filling of the organ) into cystic formations. With polycystic disease, instead of healthy functioning kidney tubules, blisters filled with fluid form. Due to the growth of cysts, the kidney gradually loses its ability to filter.
The kidney is an organ that filters the blood and removes toxins. Losing functionality, it cannot ensure the removal of toxic substances, which leads to serious pathological changes in the body, gradually leading to death. Therefore, polycystic disease requires competent and effective treatment, which only a modern clinic can provide.
For the diagnosis and treatment of polycystic kidney disease, we invite you to the Diana private clinic. You can make an appointment with a urologist at any time convenient for you.
Symptoms of polycystic kidney disease
In the initial stages, the disease is asymptomatic, so the pathology is detected by chance, most often by ultrasound.
In the presence of serious violations, the clinical picture becomes clear. The patient is tormented:
- Heaviness in the back, dull pain in the lower back; thirst;
- Frequent urination with a large volume of liquid.
- Weakness and blood in the urine are observed.
Polycystic disease in an autosomal recessive form leads to the development of chronic renal failure. This is the most dangerous complication of polycystic kidney disease. In this case, due to severe renal dysfunction, the patient experiences:
- arterial hypertension, arrhythmia;
- increase in body temperature up to 40 C, accompanied by chills;
- intestinal disorders;
- skin itching.
An infection often occurs and inflammation of the kidney develops - pyelonephritis, urolithiasis, infertility (in men). The cyst may fester and rupture. Complications significantly aggravate the course of the disease, provoking even greater disruptions in the functioning of the body.
general description
Polycystic kidney disease is a genetically determined pathology, which consists of the replacement of the renal parenchyma with cysts.
Throughout human life, polycystic dysplasia continuously progresses. It is most often diagnosed between the ages of 20 and 40, much less often in childhood and old age.
It is believed that this pathology develops as a result of an embryonic malformation of the renal tubules, when a certain number of them are converted into cysts. At the same time, the kidneys themselves increase in size due to the content of cysts of various volumes, where areas of functioning renal tissue alternate with areas of fibrosis. The pyelocaliceal system of the kidney is compressed and deformed by cysts, which can suppurate. It is characteristic that almost all patients with polycystic kidney disease can feel completely healthy for many years. Polycystic disease is discovered accidentally, for example, during an abdominal ultrasound, surgery, or even a post-mortem autopsy.
Symptoms
Polycystic kidney disease is characterized by a gradual increase in symptoms. Until a certain point, the disease seems to sleep and does not manifest itself in any way. The disease begins with severe pressing pain in the lumbar region and abdominal pain. Cysts seem to stretch the kidneys, which over time leads to dysuria and hematuria. Over time, a person begins to get tired quickly, he is often tormented by attacks of headaches, and kidney failure develops. When cysts suppurate, fever may occur, as well as symptoms of general intoxication and the development of sepsis.
Pathological anatomy
Macroscopic structure
The fetus has a characteristic appearance (Potter's face): narrow eyelid slits, a characteristic groove under the eyelid line, micrognathia, a flattened nose, soft large ears of an abnormal shape. The kidneys are increased in size, while maintaining their bean-shaped shape, but cannot perform their function. Poorly functioning kidneys do not produce enough fetal urine, leading to oligohydramnios and pulmonary hypoplasia. The hydrostatic pressure of the amniotic fluid ensures the normal development of the respiratory system.
Figure 1 | Fetus, 23 weeks. Fetal death after premature birth was due to pulmonary hypoplasia, caused by oligohydramnios due to PBP. Enlargement of both kidneys with displacement of the abdominal organs in combination with lung hypoplasia.
Figure 2 | Longitudinal section of a kidney with PCB.
Microscopy
Kidneys Radially oriented dilated collecting ducts form renal cysts with a diameter of 1–2 mm, between which normal glomeruli and tubules are visible.
The size of cysts can vary depending on age. In the early stages, the disease begins with microcysts, which then grow and turn into macrocysts. Cystic kidney disease is accompanied by slight interstitial fibrosis of the renal parenchyma.
Figure 3 | Microscopic specimen of a kidney from a patient with PBC-AR. Twenty-fold magnification, hematoxylin-eosin staining. ✱ — radial renal cysts; ▼—renal capsule. Arrows indicate normal glomeruli between dilated collecting ducts.
Liver Histological changes in the liver include the following malformations of the ductal plate: biliary hyperplasia, biliary ectasia and periportal fibrosis.
Disturbances in the morphogenesis of the development of the bile ducts lead to their dilatation. With subsequent progression of the disease, the dilated ducts turn into macrocysts connected by normal ducts, which allows them to be verified quite well using MRCP.
Figure 4 | Pathological changes in the liver. a) The structure of the normally branched portovenous and ethmoidal systems of the biliary tract (left) is disrupted due to a defect in the development of the ductal plate and errors in the terminal differentiation of cholangiocytes (right); b) Coronal T2-weighted image of the abdominal cavity: - the arrow indicates a cystic fusiform dilatation of the bile ducts; — arrow like an arrowhead — nephromegaly with small cysts; ✱ — splenomegaly. c) Preparation of a liver fragment, hematoxylin-eosin staining, 40x magnification:
✱ — extensive fibrosis of the portal area;
arrow - dilated tortuous bile ducts; arrowhead type arrow —hypoplasia of tributaries of the portal vein.
Diagnostics
The diagnosis of PKD-AR should be suspected in all cases of diffuse echogenic enlargement of both kidneys on ultrasound. Clinical signs and radiographic changes are usually sufficient to make an accurate diagnosis. But for accurate verification of the disease, special diagnostic criteria have been developed: a combination of pathognomonic signs of kidney changes - one or more of the following:
- ectasia of the bile ducts (see ultrasonography);
- clinical and laboratory signs of congenital liver fibrosis (CHF);
- hepatobiliary pathology, characteristic of anomalies in the development of the ductal plate and accompanying VFP;
- the presence of morphological (biopsy or autopsy) and genetic features.
Ultrasonography
High-resolution ultrasound has significantly improved the diagnosis of mild forms of the disease, when clinical manifestations are mild, providing non-invasive, detailed visualization of changes in the kidneys without the use of radiation or contrast agents.
Ultrasonography is the method of choice when examining the fetus and pediatric forms of PCD-AR. Ultrasound diagnostics has a large list of advantages: low cost, painlessness, widespread use and no need for radiation and sedation. But despite all the listed advantages of ultrasound, for the purpose of verification, the diagnosis is supplemented by MRI.
Pathognomonic changes in the kidneys diagnosed by ultrasound:
- increase in kidney size (taking into account age-related characteristics);
- increased echogenicity;
- decreased cortico-medullary differentiation.
In utero:
- enlarged bean-shaped kidneys with smoothed corticomedullary differentiation, oligohydramnios and an empty bladder in some cases are found;
- severe oligohydramnios leads to pulmonary hypoplasia and high mortality due to pulmonary failure. In addition, oligoamnion can be combined with multiple anomalies of the face, limbs and other organs such as Potter's syndrome.
Infancy (up to one year):
- the presence of palpable space-occupying lesions in both lateral areas of the abdomen with idiopathic chronic pulmonary pathology, a history of oligohydramnios in the mother or spontaneous pneumothorax in the newborn and arterial hypertension suggest PBP-AR;
- The combination of biliary abnormalities together with signs of portal hypertension, such as hepatosplenomegaly, adds confidence to the diagnosis of PBP-AR.
Childhood and adolescence:
- with age, the size and degree of renal fibrosis will progress along with hepatobiliary tract abnormalities, leading to progressive portal hypertension;
Figure 5 | Both kidneys are enlarged in longitudinal size (right 10 cm and left 9 cm), with increased echogenicity (due to acoustic enhancement of tiny cysts) and altered corticomedullary differentiation.
Figure 6 | Liver with congenital fibrosis and cystic extensions in the right lobe.
Figure 7 | MRI, coronal T2 projection, patient with PBP-AR.
MRI
Magnetic resonance imaging does not have any advantages over high-end ultrasound scanners and genetic testing in diagnosing PKP-AD.
Magnetic resonance cholangiopancreatography (MRCP)
MRCP images provide good visualization of the hepatobiliary system in patients with PBP-AR. MRCP is a sensitive method for diagnosing biliary ectasia, which, in combination with renal imaging, has almost completely replaced invasive diagnostic methods such as renal and liver biopsies. NB! Kidney and liver biopsies should not be used to diagnose PLD.
Figure 8 | MRCP and subsequent rendering of the biliary system with anomalies characteristic of patients with PBP-AR: A. Normal liver with an enlarged gallbladder, (S) stomach. B. Enlarged common bile duct with gallbladder. Dilated intrahepatic ducts. C. Small spindle-shaped cysts of the peripheral and central bile ducts with an enlarged common bile duct (arrow) and gallbladder. D. Fusiform macrocysts with dilated hepatic ducts. All patients had an abnormally shaped liver with a disproportionately enlarged left lobe.
Differential diagnosis
PBP-AR should be differentiated from the following conditions:
- autosomal dominant polycystic kidney disease;
- renal dysplasia associated with trisomy 13;
- Meckel-Gruber syndrome;
- Lawrence-Moon-Bardet-Biedl syndrome;
- Beckwith-Wiedemann syndrome;
- glomerular cystic (glomerulocystic) kidney disease.
Diagnosis confirmation
If PBP-AR is strongly suspected, genetic testing and counseling are necessary. Molecular genetic testing is indicated for a proband with cystic enlargement of the kidneys and VFP to identify both pathological alleles of PKHD1. At least 3 generations must be analyzed, since the disease is transmitted recessively. Molecular genetic testing approaches include single point mutation panels, multigene panels, or whole genomic testing.
What diseases can it be associated with?
Cerebral aneurysms are protrusions of the walls of arteries (less often veins) of the brain due to their thinning or stretching. The pathology often accompanies polycystic kidney disease. The consequence of rupture of a cerebral aneurysm is subarachnoid or intracerebral hemorrhage, and this is dangerous for neurological disorders of varying severity and even death.
Hematuria is the presence of uncharacteristic impurities in the urine, in this case blood, which can be noticeable to the human eye (macrohematuria) or detected by microscopic examination (microhematuria).
Hypertension is a persistent increase in blood pressure caused by compression of the renal artery, which subsequently affects the general blood flow. There is an excessive production of substances that regulate blood pressure.
Colon diverticulosis is a small but multiple expansion of the intestinal wall, which affects its elasticity and is associated with constipation. Pathology can often accompany polycystic kidney disease.
Pancreatic cysts are abnormally formed cavities in the pancreatic tissue, which contain pancreatic secretions and tissue detritus. Pathology can often accompany polycystic kidney disease.
Metabolic disorders are a consequence of the degeneration of kidney tissue, which interferes with normal metabolism, in particular, the body is oversaturated with nitrogenous substances and intoxication occurs.
Hereditary disorders are recognized as the root cause of the development of polycystic kidney disease.
Pyelonephritis is an inflammation of predominantly bacterial origin, affecting the glomeruli of the kidney system, characterized by damage to the renal pelvis, calyces and renal parenchyma. It is highly likely to develop in patients with polycystic kidney disease, since multiple cysts change the structure of the kidneys and make them more susceptible to bacterial attack.
Mitral valve prolapse is a disease characterized by dysfunction of the valve located between the left atrium and the ventricle. It is often a concomitant disease with polycystic kidney disease.
Polycystic ovary syndrome is a polyendocrine syndrome that combines dysfunction of the ovaries (absence or irregularity of ovulation, increased secretion of androgens and estrogens), pancreas (hypersecretion of insulin), adrenal cortex (hypersecretion of adrenal androgens), hypothalamus and pituitary gland.
Chronic renal failure is a slowly progressive kidney disease, dangerous due to general intoxication of the body due to years of developing imbalance of water-salt and acid-base metabolism. It is the final stage of polycystic kidney disease.
Peculiarities
As a rule, polycystic kidney disease is an embryonic defect that occurs in the tubules of these organs and is characterized by the formation of multiple small cysts on the renal parenchyma. When identifying pathology, it is necessary to remember that this is a bilateral disease that affects both kidneys. Cysts come in different sizes - from a few millimeters to several centimeters. They are light or chocolate in color and produce a jelly-like secretion.
However, over time, the size of the cysts steadily increases, resulting in a progressive decrease in parenchymal tissue, which becomes the main cause of the development of renal failure. Polycystic disease can also affect other organs: the pancreas, spleen, liver and seminal vesicles.
Classification
Depending on the time of onset of the first manifestations and the degree of liver damage, the following types of disease are distinguished:
1. Perinatal type - the most common:
- accompanied by oligohydramnios and pulmonary hypoplasia;
- 75% die within 24 hours of birth;
- minimal degree of liver fibrosis;
2. Neonatal type - minimal degree of periportal liver fibrosis;
3. Infantile type - moderate periportal fibrosis;
4. Juvenile type - pronounced periportal fibrosis in combination with portal hypertension, splenomegaly and portosystemic varicose veins.