Болезнь Фабри или болезнь Андерсона-Фабри – наследственное заболевание, относящееся к группе лизосомных болезней накопления, обусловленное значительным снижением активности или отсутствием фермента α-галактозидазы А. Дефицит фермента приводит к накоплению глоботриаозилцерамида и родственных гликофосфолипидов в лизосомах клеток различных органов, включая сердце, почки, нервную систему и эндотелий сосудов. Впервые болезнь Фабри была описана немцем Йоханнесом Фабри и англичанином Уильямом Андерсоном независимо друг от друга. В 1898 году Фабри описал 13-летнего мальчика с нодулярной пурпурой и последовавшей за ней альбуминурией. Он классифицировал данные симптомы как вариант диффузной ангиокератомы. В этом же году Андерсон описал 39-летнего мужчину с ангиокератомой, протеинурией, деформациями пальцев рук, варикозным расширением вен и лимфоотеком . Андерсон предположил наличие мультисистемного заболевания. Более распространенное название болезни – болезнь Фабри, другой вариант названия – болезнь Андерсона-Фабри.
Этиология
Причиной возникновения болезни Фабри являются мутации гена GLA, контролирующего структуру α-галактозидазы А. Альфа-галактозидаза — фермент, образующийся в недостаточном количестве при синдроме Фабри. Болезнь Фабри наследуется по рецессивному X-сцепленному типу. Гемизиготные мужчины имеют единственную мутантную Х-хромосому, что определяет классический фенотип болезни. Они передают мутантную хромосому только своим дочерям, но не сыновьям. Таким образом дочери больных болезнью Фабри отцов имеют одну нормальную и одну мутантную хромосому, т.е. являются гетерозиготами. Оставаясь, как правило, клинически здоровыми, они могут передать мутантную хромосому и, следовательно, патологический аллель половине своим потомкам. Течение болезни у них, как правило, умеренно-выраженное с более поздним началом, медленным прогрессированием и легкими клинико-патологическими изменениями. Вместе с тем было показано, что у части гетерозиготных женщин с мутацией гена α-галактозидазы А развиваются тяжелые проявления болезни Фабри, требующие медицинской помощи и вмешательства. Механизм, посредством которого у гетерозиготных женщин развиваются жизнеугрожающие симптомы, неизвестен. У большинства из них имеется почти нормальный уровень циркулирующего фермента за счет того, что случайный процесс инактивации Х-хромосомы (лайонизация) приводит к образованию как дефицитных, так и нормальных клеток. Таким образом, гетерозиготных женщин не следует называть носителями, поскольку носительство подразумевает отсутствие клинических проявлений болезни Фабри. Все дочери мужчины с болезнью Фабри и здоровой женщины будут носительницами, а один из сыновей имеет шанс быть здоровым. Источник: https://old.icahn.mssm.edu/research/programs/international-center-for-fabry-disease/fabry-disease Гликосфинголипиды, такие как глоботриаозилцерамид и галабиозилцерамид, имеют терминальный остаток α-галактозила, который отщепляется ферментом α-галактозидазой А. Таким образом, недостаточность или отсутствие фермента приводит к накоплению различных гликосфинголипидов с терминальным α-галактозил остатком.
Патогенез
Первичным патогенетическим звеном болезни Фабри является дефицит α-галактозидазы А, которая в норме отщепляет терминальный остаток α-галактозы олигосахаридной цепи нейтральных гликосфинголипидов. Недостаточность фермента приводит к накоплению в лизосомах церамидтригексозида (эндотелиальные и гладкомышечные клетки сосудов, эпителиальные и перителиальные клетки большинства органов) и дигалактозилцерамида (почки, камеры сердца, центральная нервная система). Считают, что во многих органах, таких как сосудистый эндотелий, мышцы и почки, депонирование гликосфинголипидов вызывается повышенным уровнем циркулирующих липидов, которые накапливаются путем активного всасывания и диффузии. В нервной системе также происходит накопление глоботриаозилцерамида через проницаемый гематоэнцефалический барьер.
Классификация
Выделяют две формы болезни – классическую и атипичную (позднее начало, изолированное поражение головного мозга, сердца или почек). В англоязычной литературе также выделяется «женский» вариант развития болезни. Первые симптомы болезни обычно появляются в подростковом возрасте. В большинстве случаев у мужчин заболевание протекает более тяжело. Обычно у женщин первые симптомы заболевания появляются на 5-10 лет позднее, чем у мужчин (Mehta & Ginsberg, 2005). Пациенты с БФ имеют высокий риск смертельного исхода от почечных, сердечно-сосудистых и цереброваскулярных осложнений (Colombi et al., 1967; MacDermot et al., 2001a; MacDermot et al., 2001b).
Кто наследует недуг?
Многие люди задаются вопросом о том, как проявляется болезнь Фабри, что это такое? Согласно проведенным исследованиям, медики пришли к таким выводам:
- Заболевание может развиваться у человека вне зависимости от половой принадлежности.
- Ген, обуславливающий развитие болезни, локализуется в X-хромосоме. У мужчин есть только одна X-хромосома, и если она содержит пораженный ген, вероятность развития симптоматики очень велика. Мужчина не может передать болезнь сыновьям, но все дочери наследуют этот дефект.
- Женщины имеют две X-хромосомы, поэтому вероятность передачи недуга детям любого пола составляет 50/50 %.
- Симптомы часто проявляются в детском и подростковом возрасте, а также у молодых людей.
- Дефектный ген встречается у одного из 12 000 новорожденных, то есть нечасто.
Проявления
Клинические проявления болезни Фабри в разных возрастных группах у мужчин и женщин (результаты анализа базы данных FOS). Мужчины – голубые столбцы, женщины – темно-синие. Опубликовано с разрешения Mehta et al. (2004) Периферическая нервная система:
- В 70% случаев выраженные и изнуряющие нейропатические боли. Они могут иметь как хроническое, так и кризовое течение, обычно возникают в подростковом возрасте. В ряде случаев они являются первыми симптомами заболевания и могут начинаться очень рано – от 2-х лет.
- Акропарестезии – мучительные, жгучие боли, которые больные ощущают преимущественно в ладонях и подошвах стоп, иррадиирующие в проксимальные отделы конечностей.
- Болевые кризы часто возникают при перемене погоды и лихорадке.
- Болевой синдром при БФ может усугубляться при физической нагрузке и стрессе. В ряде случаев описано снижение температурной чувствительности.
- Иногда необходима диф. диагностика с ревматическими болями.
- В ряде случаев боли могут иррадиировать в мышцы или в область живота, иметь острый, колющий характер, имитируя картину острого аппендицита или почечной колики.
- Иногда боли столь изнуряющие и мучительные, что приводят к суицидальным попыткам.
Точный механизм нейропатической боли при БФ все еще неизвестен. Предполагается, что боль является результатом структурных повреждений нервных волокон в результате накопления Gb3 в аксонах нервов, задних корешках спинномозговых ганглиев и vasa nervorum. Усиление болевого синдрома при снижении температуры при этом вызвано сужением просвета сосудов и нарушениями микроциркуляции, что приводит к нарушению трофики нервных волокон. Считается, что в поврежденных нервных волокнах происходит увеличение числа кальциевых каналов, что приводит образованию патологических симпатических афферентных связей и растормаживанию путей ноцицептивной чувствительности как центральной, так и периферической нервной системы. Гипогидроз / гипергидроз
- часто встречается снижение или полное отсутствие потоотделения.
- В некоторых случаях повышенное потоотделение.
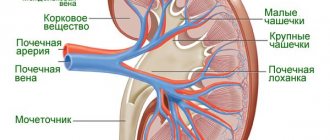
Эти изменения связаны с накоплением Gb3 в потовых железах и в стенках кровоснабжающих их сосудов. Под действием нарушения потоотделения и нейропатических болей, у детей и подростков снижается переносимость физических нагрузок. В жаркую погоду больные особенно часто предъявляют жалобы на головокружение, головную боль, тошноту, диспноэ, возможна утрата сознания. Снижение потоотделения часто сочетается со снижением слезоотделения и саливации. Центральная нервная система У пациентов с БФ высокий риск развития ишемических инсультов (встречаются в 27% случаев, часто в молодом возрасте). Основными симптомами цереброваскулярного поражения являются гемипарезы, дизартрия, нистагм, двоение в глазах и нарушения концентрации внимания. Исследования головного мозга пациентов с БФ показали, что природа этих нарушений – патология сосудов (извитость и расширение). Преходящие нарушения мозгового кровообращения могут возникать в любых сосудистых бассейнах головного мозга с преимущественной локализацией в задних отделах, что объясняется их повышенной перфузией. Инсульты могут развиваться у пациентов с не неврологической манифестацией болезни или обусловливаться накоплением Gb3 в церебральных сосудах, изменениями локального кровотока или, возможно, нарушениями функцией сосудов. Также возможен односторонний или двусторонний шум в ушах, головокружения и головные боли, а также интеллектуальный дефицит, нарушения поведения и снижение слуха. Почечная патология Ранний симптомом поражения почек, микроальбуминурия – экскреция альбумина с мочой от 30 до 300 мг/24 часа (20-200 мкг/мин за ночь) или соотношение альбумин/креатинин от 2,5 до 25 мг/ммоль (альбумин 20-200 мг/л). В развернутой стадии БФ с поражением почек наблюдается макроальбуминурия (протеинурия) – уровень экскреции альбумина выше 300 мг/24 часа (выше 200 мг/мин за ночь) или соотношение альбумин/ креатинин выше 25 мг/ммоль (альбумин выше 200 мг/л). Возможно обнаружение в моче Gb3, но при «неклассическом» варианте БФ у мужчин или классическом у женщин он может отсутствовать. Необходим дифференциальный диагноз с гомоцистинурией, которая тоже приводит к развитию сосудистых нарушений. Для гомоцистинурии существует специфическое лечение (витамин В6, фолиевая кислота и бетаин). Недавние исследования показали, что у пациентов с БФ могут возникать кисты в почках, располагающиеся вокруг почечных лоханок. Сердечные нарушения Частым симптомом при БФ является поражение сердца в результате структурных и функциональных изменений в миокарде, проводящей системе и клапанах сердца. Отложение Gb3 обнаруживают во всех структурах сердца: миокард, эндокард, эндотелий, проводящая система сердца. Отложения Gb3 выявляют и в кровеносных сосудах легких. Дебют заболевания может проявляться различными сердечными симптомами: стенокардией, диспноэ, болевым синдромом в области сердца, учащенным сердцебиением и синкопальными состояниями. Клиническая симптоматика со стороны сердца преимущественно обусловлена развитием прогрессирующей гипертрофической кардиомиопатии. Прогрессирующая концентрическая гипертрофия левого желудочка (утолщение стенок сердца без значительной дилатации полости левого желудочка) является наиболее частым нарушением при БФ Практически у всех мужчин с классической формой БФ гипертрофическая кардиомиопатия развиваетсяк 30, а у больных женщин – к 40 годам. Выраженность кардиомиопатии при БФ зависит от возраста и остаточной активности a-галактозидазы А. МРТ сердца пациента с болезнью Фабри (систола). Полость левого желудочка не визуализируется вследствие концентрической гипертрофии миокарда. Препарат сердца при БФ. Рестриктивная кардиомиопатия. Повышенное артериальное давление не играет ключевой роли в развитии заболевания. Многие авторы считают, что БФ, манифестирующая изолированной гипертрофической кардиомиоптией, часто пропускается. Механизмы, отвечающие за увеличение левого желудочка при БФ остаются до конца не выясненными, поскольку Gb3 составляет только малую часть (примерно 1%) от общей массы левого желудочка. Кожные изменения Ангиокератомы (диффузная ангиокератома туловища) — небольшого размера поверхностные ангиомы, возвышающиеся над поверхностью кожи, темно-красного цвета. Ангиокератомы обычно появляются в подростковом возрасте, по данным FOS они встречаются у 50% мужчин и 41% женщин младше 18 лет. Располагаются на поверхности тела группами и занимают большую поверхность тела. В некоторых случаях встречаются единичные ангиокератомы, которые можно увидеть только при тщательном клиническом осмотре больного. Ангиокератомы могут встречаться в любой части тела с преимущественной локализацией на бедрах, ягодицах, в паховых областях, нижней части живота и половых органах, могут наблюдаться на слизистых, таких как конъюнктива, ротовая полость, верхние дыхательные пути, желудочно-кишечный и мочеполовой тракты. Ангиокератомы при болезни Фабри. (а) типичная ангиокератома в области бедер и ягодиц; (б) ангиокератома в области пупка; (в) ангикератома губ; (г) ангиокератома на ладонях Офтальмологические нарушения
- «мутовчатое» помутнение роговицы (cornea verticillata), которое встречается у большинства пациентов с данным заболеванием. «Мутовчатое» помутнение роговицы представляет собой билатеральные изменения по типу пучка листьев или цветков на конце стебля, расположенные в поверхностном слое радужки
Cornea verticillata Cornea verticillata Cornea verticillata
- наблюдаются два характерных типа помутнения хрусталика: двусторонняя передняя капсулярная или подкапсулярная катаракта, часто имеющая «пропеллер-подобное» распределение с радиальным распространением и радиальная задняя субкапсулярная катаракта, которая очень специфична для БФ (катаракта «Фабри»). Катаракта Фабри представляет собой линейные, спицеобразные, радиально направленные отложениея гранулярного материала (беловатого и полупрозрачного) локализованного на или рядом с задней капсулой хрусталика.
Задняя субкапсулярная катаракта (спицеообразная катаракта, катаракта Фабри)
- У некоторых больных отмечается повышенная извитость сосудов конъюнктивы. Отложения гликосфинголипидов находят в эндотелиальных клетках, перицитах, гладкомышечных клетках сосудистой оболочки конъюнктивы.
Извитые сосуды конъюнктивы
- На поздних стадиях заболевания может появиться отек век.
Нарушения слуха У большинства пациентов с БФ наблюдается прогрессирующее снижение слуха. Чаще развивается нейросенсорная тугоухость, в некоторых случаях в сочетании с кондуктивной. Как правило, наблюдается постепенное снижение слуха, но иногда отмечается острая потеря слуха. Приблизительно у 85% мужчин к 50 годам и у 75% женщин к 60 годам наблюдается тяжелая тугоухость, что требует ношение слухового аппарата. Шум в ушах при БФ отмечается намного чаще, чем в популяции. Желудочно-кишечные расстройства Желудочно-кишечные расстройства встречаются в 50-70% случаях. Общие гастроинтестинальные симптомы:
- диарея,
- снижение перистальтики кишечника в сочетании со схваткообразными болями в животе
- метеоризмы,
- позывы к частым мочеиспусканиям и вздутие живота вследствие скопления газов в кишечнике.
- После приема пищи могут возникать тошнота, рвота, ощущение быстрого насыщения и боли в эпигастральной области.
- Вышеуказанные симптомы часто приводят к снижению аппетита и уменьшению частоты приемов пищи в сутки. У таких пациентов возникает дефицит массы тела и гипотрофия.
- У многих пациентов эпизоды диареи сменяются периодами нормального стула или запорами; указанные противоположные симптомы объясняют синдромом раздраженного кишечника, боли в животе и вздутие, появляющиеся после приема пищи.
- Назначение церукала (метоклопрамида) при БФ улучшает перистальтику.
- Более редкими желудочно-кишечными симптомами являются анорексия, ахалазия, дивертикулез тощей кишки с перфорацией, кровавая рвота (гематомезис) с эктазией пищевода. Иногда встречается недостаточность поджелудочной железы.
- У некоторых пациентов в результате частых запоров развивается геморрой.
Патогенез желудочно-кишечных симптомов остается до конца не выясненным, одним из механизмов считается накопление Gb3 в нейронах подслизистых и мышечных нервных сплетений, приводящее к тонкокишечной нейропатии. Дыхательные нарушения Обструкция верхних дыхательных путей является поздним осложнением БФ и может быть очень тяжелой. В двух исследованиях, изучающих функции дыхательных путей у пациентов с диспноэ, было показано, что у больных снижается объем форсированного выдоха в 1, и чаще отмечаются инфекции дыхательных путей. Обструкция дыхательных путей более ярко выражена у пожилых пациентов с БФ. Другие проявления болезни Фабри У многих гемизиготных мужчин выявлена задержка полового развития и скудное оволосение на лице и теле. В некоторых семьях болезнь симулирует симптомы акромегалии (разрастание мягких тканей, выступающая нижняя челюсть и лобные бугры, запавшая переносица). Большинство пациентов с БФ имеют мышечно-скелетные аномалии. У пациентов с БФ часто встречается анемия, которая является дополнительным фактором, осложняющим состояние пациентов с поражением почек, инсультами, сердечной недостаточностью. Гипотиреоз, остеопения также описаны у больных с БФ. Искажения черт лица, характерные для больного синдромом Фабри. Пациент был подвержен фермент-заместительной терапии на протяжении десяти лет, у него ярко выраженные надбровные дуги, густые брови, мешки под глазами, неполный блефароптоз (опущение века), прогения. Лицевые аномалии разрешаются по ходу лечения ферментативной заместительной терапией.
Пренатальная диагностика в немецких клиниках
Выявление и лечение болезни Фабри в Германии начинают уже на внутриутробном этапе развития. Пренатальная диагностика возможна следующими методами:
- определение активности альфа-галактозидазы;
- ДНК-анализ ворсин хориона;
- исследование амниотической жидкости;
- анализ крови плода.
Определение диагноза болезнь Фабри на генетическом уровне проводят молекулярно-генетическими и биохимическими методами. Положительный результат диагностики требует последующего обследования всей семьи.
Заключительный диагноз выставляется на основе симптомов и уровне активности галактозидазы в следующих средах:
- тканевых биоптатах;
- культивируемых фибробластах;
- слезной жидкости;
- плазме и моче.
Дифференцируют болезнь Фабри с телеангиэктазиями и ревматическими поражениями органов. Пренатальная диагностика – основной способ предупреждения болезни у ребенка.
Терапия пациентов с диагнозом «болезнь Фабри» проводится консервативными методами. Назначают препараты, предупреждающие кризы: дифенилгидантоин, карбамазепин. На поздних стадиях выполняют пересадку почки.
Лечение болезни Фабри
В настоящее время применяют два подхода к лечению этого заболевания – симптоматическое лечение и ферментную заместительную терапию (α-галактозидаза А).
Симптоматическое лечение
Лечение болевого синдрома
Самым ранним, мучительным и снижающим качество жизни симптомом при БФ являются боли. По рекомендации ВОЗ для лечения хронической невропатической боли применяют мембраностабилизаторы, такие как габапентин, карбамазепин или фенитоин. Также могут применяться антидепрессанты, например амитриптилин. При необходимости, во время температурных кризов, к лечению присоединяют нестероидные противовоспалительные средства или опиаты – инфузии морфина в низких дозах. К болевому синдрому у детей при БФ и другим симптомам, как правило, не относятся серьезно. Необходимо разъяснять учителям необходимость снижения физической активности для пациентов с БФ, поскольку физическая активность является провоцирующим фактором развития болевых кризов. Взрослые с БФ должны получить рекомендации по выбору их профессиональной деятельности. Почечная патология при БФ Антигипертензивные средства и препараты, защищающие почечную паренхиму, должны назначаться после появления признаков поражения почек. При прогрессирующей почечной недостаточности требуется гемодиализ и в некоторых случаях трансплантация почки. Не рекомендуется в качестве доноров почки привлекать родственников пациентов с БФ или женщин, гетерозиготных носительниц. Сердечно-сосудистые нарушения У пациентов с выраженной брадикардией необходимо установить водитель ритма. Больным с тахикардией, как с установленным водителем ритма так и без такового, должны быть назначены антиаритмические препараты. В случае если у пациентов с БФ наблюдается аритмия, резистентная к терапии, назначают антикоагуляционную терапию. Некоторые авторы рекомендуют назначение антиагрегантных препаратов, например низких доз аспирина. Ангиокератомы Если пациент решил удалить ангиокератомы с косметической или какой-либо другой целью (например, постоянная кровоточивость) рекомендуют проведение лазеротерапии. Хирургическое удаление ангиокератом в связи с их диффузным распространением и неизбежным образованием рубцов в месте их удаления применяют реже. Желудочно-кишечные расстройства Соблюдение определенного режима питания: дробное питание небольшими порциями. У ряда пациентов хороший эффект наблюдался при подключении к терапии панкреатических ферментов.
Фермент-заместительная терапия
Первый успешный опыт ФЗТ был получен в 1970х годах. Для лечения использовали α-галактозидазу А, выделенную из человеческой плаценты. В то время не было возможностей проведения адекватного широкомасштабного клинического испытания, поскольку количество очищенного фермента было очень небольшим. Первыми в клиническом испытании приняли участие два брата с БФ, у которых не было отмечено побочных реакций на препарат, и которые хорошо переносили инфузии. Применение методов генной инженерии позволило достичь значительного прогресса в получении больших количеств α-галактозидазы А – фермент стали получать с применением клеточных линий человека (agalsidase alfa) и клеток яичника китайского хомяка (agalsidase betta), именно это сделало возможным лечение многих пациентов. При рандоминизированном двойном слепом клиническом испытании в одном из центров 26 пациентов с БФ получали плацебо или агалзидазу альфа (в дозе 0,2мг/кг внутривенно, длительность инфузии 20-40 мин, каждые 2 недели) в течение 6 месяцев. При применении препарата отмечалось статистически достоверное снижение интенсивности болей и улучшение качества жизни пациентов по сравнению с группой пациентов, получающих плацебо Побочные эффекты. Переносимость длительной ФЗТ хорошая. Основными нежелательными эффектами могут быть реакции на введение препарата (озноб, лихорадка, тошнота, тахикардия, зуд, миалгия, боли в конечностях, головная боль, боль в груди), в основном легкие или умеренно выраженные. В случае анафилактической реакции должна осуществляться неотложная медицинская помощь, инфузии ФЗТ должны быть приостановлены, срочно должен быть взят анализ на IgE. Когда начинать заместительную терапию?
Оптимальные сроки назначения заместительной терапии не определены, а рекомендации по назначению пациентов с болезнью Фабри отличаются в разных странах. Мужчинам с болезнью Фабри заместительная терапия показана сразу после установления диагноза. У мальчиков с бессимптомной болезнью Фабри лечение может быть начато в возрасте 10-13 лет, в то время как при наличии симптомов откладывать заместительную терапию не следует. У женщин показаниями к лечению считают выраженные симптомы или признаки прогрессирующего поражения органов-мишеней, в том числе хроническая невропатическая боль в кистях и стопах, резистентная к стандартной терапии, персистирующая протеинурия, снижение скорости клубочковой фильтрации <80 мл/мин/1,73 м2, поражение сердца, нарушения мозгового кровообращения или ишемические изменения головного мозга, выявленные при магнитно-резонансной томографии.
Генотерапия. Цель генотерапии – введение функциональной копии дефектного гена в некоторые или во все клетки организма. Успешная трансфекция гена кодирующего α-галактозидазу А в клетки костного мозга, полученных от пациентов с болезнью Фабри, была продемонстрирована в ряде исследований. Однако, несмотря на значительные успехи, применение генотерапии у людей не может быть начато в ближайшее время. Фармакологические шапероны. У некоторых пациентов с болезнью Фабри сохраняется высокая остаточная активность α-галактозидазы А, но этот фермент является нестабильным. Фармакологические шапероны –небольшие молекулы, которые связываются с ферментом и не допускают его разрушения, обеспечивая проникновение белка в эндоплазматический ретикулюм и лизосомы. В лизосомах фармакологические шапероны отсоединяются от белка и фермент начинает работать. На данный момент в мире лечение не зарегистрировано.
Государственная политика:
1 марта 2010 года Совет Федерации РФ одобрил законопроект «Об обращении лекарственных средств», предусматривающий государственное регулирование цен на лекарства, относящиеся к категории жизненно необходимых и важнейших. При этом в окончательном тексте законопроекта не было упоминания об орфанных препаратах.Закон «Об обращении лекарственных средств» вступил в силу 1 сентября 2010 года. С 1 января 2012 года вступил в силу новый закон «Об основах охраны здоровья граждан Российской Федерации», в котором одна из статей посвящена редким (орфанным) заболеваниям. Впервые с принятием нового закона в России на государственном уровне было введено понятие редких заболеваний и орфанных препаратов, дано их определение и принят критерий отнесения к редким заболеваниям. Министерство здравоохранения сформировало два перечня редких заболеваний. Один включает все заболевания, которые на основе статистики, характеризующей их распространенность на территории России, можно отнести к редким (критерий отнесения ‑ не более 10 случаев на 100 тысяч населения), вне зависимости от того, существуют ли на сегодняшний день для этих заболеваний методы лечения и адекватная лекарственная терапия. В перечень вошли 230 заболеваний. Другой перечень ‑ жизнеугрожающих и хронических прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению продолжительности жизни или к инвалидности, ‑ включает 24 заболевания. Это те заболевания, для которых в настоящее время имеется патогенетическое лечение, т.е. лечение, направленное на устранение патологического процесса, а не на симптомы. 14 июня 2012 года Федеральная антимонопольная служба (ФАС) РФ представила доклад о состоянии конкуренции в Российской Федерации за 2011 год, в котором среди прочего было предложено внести изменения в законы «Об основах охраны здоровья граждан» и «Об обращении лекарственных средств». Законопроект предполагает введение ускоренной процедуры экспертизы орфанных («сиротских») лекарственных средств в целях последующей государственной регистрации. Автор: Елена Лисицына Источники:
- https://old.www.ncbi.nlm.nih.gov/books/NBK11599/
- https://old.qjmed.oxfordjournals.org/content/103/9/641
- https://old.en.wikipedia.org/wiki/Fabry_disease
- https://old.www.patient.co.uk/doctor/anderson-fabry-disease
- https://old.lookfordiagnosis.com/mesh_info.php?term=fabry+disease&lang=1
- https://old.www.aocd.org/?page=FabryDisease
- https://old.disorders.eyes.arizona.edu/disorders/fabry-disease
- https://old.emedicine.medscape.com/article/951451-overview
- https://old.eurheartj.oxfordjournals.org/content/34/11/802
- https://old.orfans.ru/
- https://old.www.cardiomyopathy.org/
Диагностика и лечение в немецких клиниках
Если у Вас в роду есть случаи наследственных болезней, то узнать подробную информацию о диагностике и ценах, можно у консультантов Deutsche Mediziniche Union. Мы ответим на все Ваши вопросы, а также возьмем все заботы по организации поездки на себя. Вы можете связаться с нами по телефону или заполнить анкету и подать заявку в электронном виде.
Наши координаторы переведут медицинскую документацию на немецкий язык и передадут профильным немецким специалистам в клиники. На основе их рекомендаций мы подберем оптимальную для Вас программу лечения и диагностики. Цены на лечение болезни Фабри в Германии составляются индивидуально. Немецкие клиники оснащены лучшим в Европе оборудованием. При этом цены вполне сопоставимы со стоимостью лечения в отечественных клиниках.
Координаторы DMU составят план поездки, позаботятся о проездных документах. Цена медицинских услуг зависит от диагностической и лечебной программы и включает предоперационную подготовку, расходы на операцию, медикаменты, послеоперационное наблюдение врача и пребывание в стационаре.
Наши координаторы оформят медицинское приглашение и визу. В Германии Вас встретит наш представитель. Мы организуем сопровождение на консультации профессиональным переводчиком. Наши специалисты проверят соответствие стоимости медицинских услуг согласно немецкому законодательству. Это поможет избежать ненужных затрат.
После лечения мы поддерживаем связь с Вами и лечащим врачом в Германии, а также позаботимся о доставке лекарства из Германии.