Fabry disease or Anderson-Fabry disease is a hereditary disease belonging to the group of lysosomal storage diseases, caused by a significant decrease in the activity or absence of the enzyme α-galactosidase A. Enzyme deficiency leads to the accumulation of globotriaosylceramide and related glycophospholipids in the lysosomes of cells of various organs, including the heart, kidneys, nervous system and vascular endothelium. Fabry disease was first described by the German Johannes Fabry and the Englishman William Anderson independently of each other. In 1898, Fabry described a 13-year-old boy with nodular purpura and subsequent albuminuria. He classified these symptoms as a variant of diffuse angiokeratoma. That same year, Anderson described a 39-year-old man with angiokeratoma, proteinuria, finger deformities, varicose veins, and lymphoedema. Anderson suggested the presence of a multisystem disease. The more common name for the disease is Fabry disease, another variant of the name is Anderson-Fabry disease.
Etiology
Fabry disease is caused by mutations in the GLA gene, which controls the structure of α-galactosidase A. Alpha-galactosidase is an enzyme produced in insufficient quantities in Fabry syndrome. Fabry disease is inherited in an X-linked recessive manner. Hemizygous males have a single mutant X chromosome, which determines the classic phenotype of the disease. They pass on the mutant chromosome only to their daughters, but not to their sons. Thus, daughters of fathers with Fabry disease have one normal and one mutant chromosome, i.e. are heterozygotes. While remaining, as a rule, clinically healthy, they can pass on the mutant chromosome and, consequently, the pathological allele to half of their descendants. The course of their disease is usually moderate with a later onset, slow progression and mild clinicopathological changes. However, it has been shown that some heterozygous women with a mutation in the α-galactosidase A gene develop severe manifestations of Fabry disease, requiring medical care and intervention. The mechanism by which heterozygous women develop life-threatening symptoms is unknown. Most of them have almost normal levels of circulating enzyme due to the fact that the random process of inactivation of the X chromosome (lyonization) leads to the formation of both deficient and normal cells. Thus, heterozygous women should not be called carriers, since carrier status implies the absence of clinical manifestations of Fabry disease. All daughters of a man with Fabry disease and a healthy woman will be carriers, and one of the sons has a chance to be healthy. Source: https://old.icahn.mssm.edu/research/programs/international-center-for-fabry-disease/fabry-disease Glycosphingolipids such as globotriaosylceramide and galabiosylceramide have a terminal α-galactosyl residue that is cleaved by the enzyme α -galactosidase A. Thus, deficiency or absence of the enzyme leads to the accumulation of various glycosphingolipids with a terminal α-galactosyl residue.
Pathogenesis
The primary pathogenetic link of Fabry disease is a deficiency of α-galactosidase A, which normally cleaves off the terminal α-galactose residue of the oligosaccharide chain of neutral glycosphingolipids. Enzyme deficiency leads to the accumulation of ceramide trihexoside (endothelial and vascular smooth muscle cells, epithelial and perithelial cells of most organs) and digalactosylceramide (kidneys, heart chambers, central nervous system) in lysosomes. In many organs, such as the vascular endothelium, muscle and kidney, glycosphingolipid deposition is thought to be caused by increased levels of circulating lipids that accumulate through active absorption and diffusion. In the nervous system, globotriaosylceramide also accumulates across the permeable blood-brain barrier.
Classification
There are two forms of the disease - classic and atypical (late onset, isolated damage to the brain, heart or kidneys). In the English-language literature, the “female” variant of the development of the disease is also highlighted. The first symptoms of the disease usually appear in adolescence. In most cases, the disease is more severe in men. Typically, women experience symptoms 5–10 years later than men (Mehta & Ginsberg, 2005). Patients with FD are at high risk of death from renal, cardiovascular and cerebrovascular complications (Colombi et al., 1967; MacDermot et al., 2001a; MacDermot et al., 2001b).
Who inherits the disease?
Many people wonder how Fabry disease manifests itself, what is it? According to the studies, doctors came to the following conclusions:
- The disease can develop in a person regardless of gender.
- The gene that causes the development of the disease is localized on the X chromosome. Men have only one X chromosome, and if it contains the affected gene, the likelihood of developing symptoms is very high. A man cannot pass the disease on to his sons, but all daughters inherit this defect.
- Women have two X chromosomes, so the chance of transmitting the disease to children of either sex is 50/50%.
- Symptoms often appear in childhood, adolescence, and young adults.
- The defective gene occurs in one in 12,000 newborns, which means it is rare.
Manifestations
Clinical manifestations of Fabry disease in different age groups in men and women (results of analysis of the FOS database). Men are blue columns, women are dark blue. Published with permission from Mehta et al. (2004) Peripheral nervous system:
- In 70% of cases there is severe and debilitating neuropathic pain. They can have both a chronic and crisis course, usually occurring in adolescence. In some cases, they are the first symptoms of the disease and can begin very early - from 2 years.
- Acroparesthesia is a painful, burning pain that patients feel primarily in the palms and soles of the feet, radiating to the proximal limbs.
- Pain crises often occur with changes in weather and fever.
- Pain syndrome in FD can be aggravated by physical activity and stress. In some cases, a decrease in temperature sensitivity has been described.
- Sometimes a differential is needed. diagnosis of rheumatic pain.
- In some cases, pain can radiate to the muscles or to the abdominal area, have an acute, stabbing nature, simulating the picture of acute appendicitis or renal colic.
- Sometimes the pain is so debilitating and painful that it leads to suicide attempts.
The exact mechanism of neuropathic pain in FD is still unknown. Pain is hypothesized to result from structural damage to nerve fibers resulting from accumulation of Gb3 in nerve axons, dorsal roots of the dorsal root ganglia, and vasa nervorum. An increase in pain with a decrease in temperature is caused by a narrowing of the lumen of blood vessels and microcirculation disorders, which leads to disruption of the trophism of nerve fibers. It is believed that in damaged nerve fibers there is an increase in the number of calcium channels, which leads to the formation of pathological sympathetic afferent connections and disinhibition of the nociceptive sensitivity pathways of both the central and peripheral nervous systems. Hypohidrosis/hyperhidrosis
- decreased or complete absence of sweating is common.
- In some cases, increased sweating.
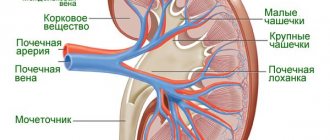
These changes are associated with the accumulation of Gb3 in the sweat glands and in the walls of the blood vessels supplying them. Due to impaired sweating and neuropathic pain, exercise tolerance in children and adolescents decreases. In hot weather, patients especially often complain of dizziness, headache, nausea, dyspnea, and possible loss of consciousness. Decreased sweating is often combined with decreased lacrimation and salivation. Central nervous system Patients with FD have a high risk of developing ischemic strokes (occur in 27% of cases, often at a young age). The main symptoms of cerebrovascular disease are hemiparesis, dysarthria, nystagmus, double vision and difficulty concentrating. Studies of the brain of patients with FD have shown that the nature of these disorders is vascular pathology (tortuosity and dilation). Transient disorders of cerebral circulation can occur in any vascular areas of the brain with a predominant localization in the posterior sections, which is explained by their increased perfusion. Strokes may develop in patients with non-neurological manifestations of the disease or be caused by accumulation of Gb3 in cerebral vessels, changes in local blood flow, or possibly impaired vascular function. Unilateral or bilateral tinnitus, dizziness and headaches are also possible, as well as intellectual deficits, behavioral disturbances and hearing loss. Renal pathology An early symptom of kidney damage, microalbuminuria is urinary albumin excretion from 30 to 300 mg/24 hours (20-200 mcg/min per night) or albumin/creatinine ratio from 2.5 to 25 mg/mmol (albumin 20-200 mg/l). In the advanced stage of FD with kidney damage, macroalbuminuria (proteinuria) is observed - the level of albumin excretion is higher than 300 mg/24 hours (above 200 mg/min per night) or the albumin/creatinine ratio is higher than 25 mg/mmol (albumin is higher than 200 mg/l). It is possible to detect Gb3 in urine, but in the “non-classical” version of FD in men or the classic version in women, it may be absent. A differential diagnosis is necessary with homocystinuria, which also leads to the development of vascular disorders. There are specific treatments for homocystinuria (vitamin B6, folic acid and betaine). Recent studies have shown that patients with FD may develop kidney cysts located around the renal pelvis. Cardiac disorders A common symptom of FD is damage to the heart as a result of structural and functional changes in the myocardium, conduction system and heart valves. Gb3 deposition is found in all structures of the heart: myocardium, endocardium, endothelium, cardiac conduction system. Gb3 deposits are also detected in the blood vessels of the lungs. The onset of the disease can be manifested by various cardiac symptoms: angina pectoris, dyspnea, pain in the heart region, rapid heartbeat and syncope. Clinical symptoms of the heart are mainly due to the development of progressive hypertrophic cardiomyopathy. Progressive concentric hypertrophy of the left ventricle (thickening of the walls of the heart without significant dilatation of the left ventricular cavity) is the most common disorder in FD. In almost all men with the classic form of FD, hypertrophic cardiomyopathy develops by age 30, and in affected women by age 40. The severity of cardiomyopathy in FD depends on age and residual activity of a-galactosidase A. MRI of the heart of a patient with Fabry disease (systole). The left ventricular cavity is not visualized due to concentric myocardial hypertrophy. Heart drug for FD. Restrictive cardiomyopathy. High blood pressure does not play a key role in the development of the disease. Many authors believe that FD manifesting as isolated hypertrophic cardiomyopsia is often missed. The mechanisms responsible for left ventricular enlargement in FD remain unclear, since Gb3 constitutes only a small portion (approximately 1%) of the total left ventricular mass. Skin changes Angiokeratomas (diffuse angiokeratoma of the trunk) are small superficial angiomas, rising above the surface of the skin, dark red in color. Angiokeratomas usually appear during adolescence, according to FOS they occur in 50% of men and 41% of women under 18 years of age. They are located on the surface of the body in groups and occupy a large surface of the body. In some cases, there are single angiokeratomas, which can only be seen with a thorough clinical examination of the patient. Angiokeratomas can occur in any part of the body with a predominant localization on the thighs, buttocks, groin areas, lower abdomen and genitals, and can be observed on mucous membranes such as the conjunctiva, oral cavity, upper respiratory tract, gastrointestinal and genitourinary tracts. Angiokeratomas in Fabry disease. (a) typical angiokeratoma in the thighs and buttocks; (b) angiokeratoma in the umbilical area; (c) angikeratoma of the lips; (d) angiokeratoma on the palms Ophthalmological disorders
- “whorled” clouding of the cornea (cornea verticillata), which occurs in most patients with this disease. Whorled corneal opacities are bilateral changes in the form of a tuft of leaves or flowers at the end of a stem, located in the superficial layer of the iris
Cornea verticillata Cornea verticillata Cornea verticillata
- There are two characteristic types of lens opacities: bilateral anterior capsular or subcapsular cataracts, often having a “propeller-like” distribution with a radial distribution, and radial posterior subcapsular cataracts, which are very specific to FD (Fabry cataract). Fabry cataracts are linear, spoke-shaped, radially directed deposits of granular material (whitish and translucent) located on or adjacent to the posterior capsule of the lens.
Posterior subcapsular cataract (spoke cataract, Fabry cataract)
- Some patients have increased tortuosity of conjunctival vessels. Deposits of glycosphingolipids are found in endothelial cells, pericytes, and smooth muscle cells of the choroid of the conjunctiva.
Twisted vessels of the conjunctiva
- In the later stages of the disease, swelling of the eyelids may appear.
Hearing Impairment Most patients with FD experience progressive hearing loss. Sensorineural hearing loss develops more often, in some cases in combination with conductive hearing loss. Typically, there is a gradual decrease in hearing, but sometimes there is acute hearing loss. Approximately 85% of men by age 50 and 75% of women by age 60 have severe hearing loss requiring hearing aids. Tinnitus in FD is much more common than in the general population. Gastrointestinal disorders Gastrointestinal disorders occur in 50-70% of cases. Common gastrointestinal symptoms:
- diarrhea,
- decreased intestinal motility combined with cramping abdominal pain
- flatulence,
- the urge to urinate frequently and bloating due to the accumulation of gases in the intestines.
- After eating, nausea, vomiting, a feeling of rapid satiety and pain in the epigastric region may occur.
- The above symptoms often lead to a decrease in appetite and a decrease in the frequency of meals per day. In such patients, body weight deficiency and malnutrition occur.
- In many patients, episodes of diarrhea are followed by periods of normal bowel movements or constipation; These opposite symptoms are explained by irritable bowel syndrome, abdominal pain and bloating that appear after eating.
- Prescribing Cerucal (metoclopramide) for FD improves peristalsis.
- More rare gastrointestinal symptoms are anorexia, achalasia, jejunal diverticulosis with perforation, hematemesis with esophageal ectasia. Sometimes pancreatic insufficiency occurs.
- Some patients develop hemorrhoids as a result of frequent constipation.
The pathogenesis of gastrointestinal symptoms remains unclear; one of the mechanisms is considered to be the accumulation of Gb3 in neurons of the submucosal and muscular nerve plexuses, leading to small intestinal neuropathy. Respiratory Disorders Upper airway obstruction is a late complication of FD and can be very severe. Two studies examining airway function in patients with dyspnea found that patients had decreased forced expiratory volume 1 and had a higher incidence of respiratory tract infections. Airway obstruction is more pronounced in older patients with FD. Other manifestations of Fabry disease Many hemizygous men have delayed sexual development and sparse hair growth on the face and body. In some families, the disease simulates the symptoms of acromegaly (soft tissue overgrowth, protruding lower jaw and frontal bumps, sunken bridge of the nose). Most patients with FD have musculoskeletal abnormalities. Anemia is common in patients with FD, which is an additional factor complicating the condition of patients with kidney damage, strokes, and heart failure. Hypothyroidism and osteopenia have also been described in patients with FD. Distortions of facial features characteristic of a patient with Fabry syndrome. The patient was subject to enzyme replacement therapy for ten years, he has pronounced brow ridges, thick eyebrows, bags under the eyes, incomplete blepharoptosis (drooping eyelid), and progenia. Facial abnormalities resolve with enzyme replacement therapy.
Prenatal diagnostics in German clinics
Detection and treatment of Fabry disease in Germany begins already at the intrauterine stage of development. Prenatal diagnosis is possible using the following methods:
- determination of alpha-galactosidase activity;
- DNA analysis of chorionic villus;
- amniotic fluid examination;
- fetal blood test.
Determining the diagnosis of Fabry disease at the genetic level is carried out using molecular genetic and biochemical methods. A positive diagnostic result requires subsequent examination of the entire family.
The final diagnosis is made based on symptoms and the level of galactosidase activity in the following media:
- tissue biopsies;
- cultured fibroblasts;
- tear fluid;
- plasma and urine.
Fabry disease is differentiated from telangiectasias and rheumatic organ lesions. Prenatal diagnosis is the main way to prevent illness in a child.
Treatment of patients diagnosed with Fabry disease is carried out using conservative methods. Prescribed drugs that prevent crises: diphenylhydantoin, carbamazepine. In later stages, a kidney transplant is performed.
Treatment of Fabry disease
Currently, two approaches are used to treat this disease - symptomatic treatment and enzyme replacement therapy (α-galactosidase A).
Symptomatic treatment
Treatment of pain syndrome
The earliest, most painful and quality-decreasing symptom of FD is pain. According to WHO recommendations, membrane stabilizers such as gabapentin, carbamazepine or phenytoin are used to treat chronic neuropathic pain. Antidepressants such as amitriptyline may also be used. If necessary, during temperature crises, non-steroidal anti-inflammatory drugs or opiates are added to treatment - morphine infusions in low doses. Pain in children with FD and other symptoms are usually not taken seriously. It is necessary to explain to teachers the need to reduce physical activity for patients with FD, since physical activity is a provoking factor in the development of pain crises. Adults with FD should receive guidance regarding their career choices. Renal pathology in FD Antihypertensive drugs and drugs that protect the renal parenchyma should be prescribed after signs of kidney damage appear. With progressive renal failure, hemodialysis and, in some cases, kidney transplantation are required. It is not recommended to involve relatives of patients with FD or women who are heterozygous carriers as kidney donors. Cardiovascular disorders In patients with severe bradycardia, a pacemaker should be installed. Patients with tachycardia, both with and without an established pacemaker, should be prescribed antiarrhythmic drugs. If patients with FD have arrhythmia that is resistant to therapy, anticoagulation therapy is prescribed. Some authors recommend the use of antiplatelet drugs, such as low-dose aspirin. Angiokeratomas If the patient decides to remove angiokeratomas for cosmetic or some other purpose (for example, constant bleeding), laser therapy is recommended. Surgical removal of angiokeratomas due to their diffuse spread and inevitable scar formation at the site of their removal is used less frequently. Gastrointestinal disorders Compliance with a certain diet: split meals in small portions. In a number of patients, good effects were observed when pancreatic enzymes were added to therapy.
Enzyme replacement therapy
The first successful experience of ERT was obtained in the 1970s. α-Galactosidase A isolated from human placenta was used for treatment. At that time, it was not possible to conduct an adequate large-scale clinical trial because the amount of purified enzyme was very small. The first to participate in the clinical trial were two brothers with FD who had no adverse reactions to the drug and tolerated the infusions well. The use of genetic engineering methods has made it possible to achieve significant progress in obtaining large quantities of α-galactosidase A - the enzyme began to be obtained using human cell lines (agalsidase alfa) and Chinese hamster ovary cells (agalsidase betta), which made it possible to treat many patients. In a randomized, double-blind, single-center clinical trial, 26 patients with FD received placebo or agalsidase alfa (0.2 mg/kg IV, 20–40 min infusion, every 2 weeks) for 6 months. When using the drug, there was a statistically significant decrease in pain intensity and an improvement in the quality of life of patients compared to the group of patients receiving placebo. Side effects. Long-term ERT is well tolerated. The main undesirable effects may be reactions to the administration of the drug (chills, fever, nausea, tachycardia, itching, myalgia, pain in the limbs, headache, chest pain), mostly mild or moderate. In the event of an anaphylactic reaction, emergency medical attention should be provided, ERT infusions should be suspended, and an IgE test should be taken immediately. When to start replacement therapy?
The optimal timing of replacement therapy has not been determined, and recommendations for prescribing patients with Fabry disease vary from country to country.
For men with Fabry disease, replacement therapy is indicated immediately after diagnosis. In boys with asymptomatic Fabry disease, treatment can be started between the ages of 10 and 13 years, but replacement therapy should not be delayed if symptoms are present. In women, indications for treatment include severe symptoms or signs of progressive target organ damage, including chronic neuropathic pain in the hands and feet, resistant to standard therapy, persistent proteinuria, decreased glomerular filtration rate <80 ml/min/1.73 m2, cardiac damage, cerebrovascular accidents or ischemic changes in the brain identified by magnetic resonance imaging. Gene therapy. The goal of gene therapy is to introduce a functional copy of a defective gene into some or all of the body's cells. Successful transfection of the gene encoding α-galactosidase A into bone marrow cells obtained from patients with Fabry disease has been demonstrated in a number of studies. However, despite significant advances, the use of gene therapy in humans cannot be started in the near future. Pharmacological chaperones. Some patients with Fabry disease have high residual α-galactosidase A activity, but this enzyme is unstable. Pharmacological chaperones are small molecules that bind to the enzyme and prevent its destruction, ensuring the penetration of the protein into the endoplasmic reticulum and lysosomes. In lysosomes, pharmacological chaperones are detached from the protein and the enzyme begins to work. At the moment, no treatment is registered in the world.
Public policy:
On March 1, 2010, the Federation Council of the Russian Federation approved the bill “On the Circulation of Medicines,” which provides for state regulation of prices for medicines classified as vital and essential. At the same time, there was no mention of orphan drugs in the final text of the bill. The Law “On the Circulation of Medicines” came into force on September 1, 2010. On January 1, 2012, a new law “On the fundamentals of protecting the health of citizens of the Russian Federation” came into force, in which one of the articles is devoted to rare (orphan) diseases. For the first time, with the adoption of a new law in Russia, the concept of rare diseases and orphan drugs was introduced at the state level, their definition was given and the criterion for classifying them as rare diseases was adopted. The Ministry of Health has created two lists of rare diseases. One includes all diseases that, based on statistics characterizing their prevalence in Russia, can be classified as rare (classification criterion is no more than 10 cases per 100 thousand population), regardless of whether treatment methods exist for these diseases today and adequate drug therapy. The list includes 230 diseases. Another list of life-threatening and chronic progressive rare (orphan) diseases leading to a reduction in life expectancy or disability includes 24 diseases. These are those diseases for which there is currently pathogenetic treatment, i.e. treatment aimed at eliminating the pathological process, and not at the symptoms. On June 14, 2012, the Federal Antimonopoly Service (FAS) of the Russian Federation presented a report on the state of competition in the Russian Federation for 2011, in which, among other things, it was proposed to amend the laws “On the Fundamentals of Protecting the Health of Citizens” and “On the Circulation of Medicines.” The bill envisages the introduction of an accelerated procedure for the examination of orphan (“orphan”) medicines for the purpose of subsequent state registration. Author: Elena Lisitsyna Sources:
- https://old.www.ncbi.nlm.nih.gov/books/NBK11599/
- https://old.qjmed.oxfordjournals.org/content/103/9/641
- https://old.en.wikipedia.org/wiki/Fabry_disease
- https://old.www.patient.co.uk/doctor/anderson-fabry-disease
- https://old.lookfordiagnosis.com/mesh_info.php?term=fabry+disease&lang=1
- https://old.www.aocd.org/?page=FabryDisease
- https://old.disorders.eyes.arizona.edu/disorders/fabry-disease
- https://old.emedicine.medscape.com/article/951451-overview
- https://old.eurheartj.oxfordjournals.org/content/34/11/802
- https://old.orfans.ru/
- https://old.www.cardiomyopathy.org/
Diagnostics and treatment in German clinics
If you have cases of hereditary diseases in your family, you can find out detailed information about diagnostics and prices from the consultants of the Deutsche Mediziniche Union. We will answer all your questions, and also take care of all the worries about organizing your trip. You can contact us by phone or fill out a form and submit an application electronically.
Our coordinators will translate medical documentation into German and transfer it to relevant German specialists in clinics. Based on their recommendations, we will select the optimal treatment and diagnostic program for you. Prices for treatment of Fabry disease in Germany are calculated individually. German clinics are equipped with the best equipment in Europe. At the same time, prices are quite comparable to the cost of treatment in domestic clinics.
DMU coordinators will draw up a travel plan and take care of travel documents. The price of medical services depends on the diagnostic and treatment program and includes preoperative preparation, surgery costs, medications, postoperative doctor’s supervision and hospital stay.
Our coordinators will issue a medical invitation and visa. Our representative will meet you in Germany. We will arrange for you to be accompanied during the consultation by a professional translator. Our specialists will check the compliance of the cost of medical services in accordance with German law. This will help avoid unnecessary costs.
After treatment, we keep in touch with you and your doctor in Germany, and also take care of the delivery of the medicine from Germany.