| Синдром Альпорта | |
| Эффект потери слуха от синдрома Альпорта у 13-летнего мальчика | |
| Специальность | Медицинская генетика |
Синдром Альпорта
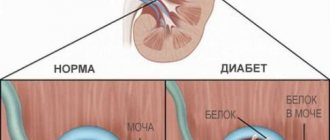
это генетическое расстройство[1] затрагивает примерно 1 из 5 000-10 000 детей,[2] был характеризован гломерулонефрит, терминальная стадия болезни почек и потеря слуха.[3] Синдром Альпорта также может влиять на глаза, хотя изменения обычно не влияют на зрение, за исключением тех случаев, когда изменения в хрусталике происходят в более позднем возрасте. Кровь в моче универсален. Протеинурия это признак прогрессирования заболевания почек.
Заболевание впервые было обнаружено в британской семье врачом. Сесил А. Альпорт в 1927 г.[4][5] Синдром Альпорта когда-то тоже имел ярлык наследственный нефрит
, но это вводит в заблуждение, поскольку существует множество других причин наследственного заболевания почек и нефрита.
Синдром Альпорта вызван наследственным дефектом коллаген IV типа- конструкционный материал, необходимый для нормального функционирования различных частей тела. Поскольку коллаген IV типа находится в ушах, глазах и почках, это объясняет, почему синдром Альпорта поражает различные, казалось бы, несвязанные части тела (уши, глаза, почки и т. Д.).
Содержание
- 1 Признаки и симптомы 1.1 Гематурия и протеинурия
- 1.2 Потеря слуха
- 1.3 Лейомиоматоз
- 1.4 Изменения глаз
- 1.5 Другие отклонения
- 2.1 Генетика 2.1.1 Шаблоны наследования
- 3.1 Биопсия почек или кожи
- 4.1 Заболевания почек и почечная недостаточность
Признаки и симптомы
Эти описания относятся к «классическому» синдрому Альпорта, который обычно вызывает серьезные заболевания у молодых людей или в позднем детстве.[6] У некоторых людей, обычно с более легкими мутациями или статусом «носителей», болезнь развивается позже или проявляются лишь некоторые черты классической болезни.
Гематурия и протеинурия
Кровь в моче — обычная черта синдрома Альпорта с раннего детства, которую можно идентифицировать щупы для измерения мочи. У маленьких детей эпизоды видимого (макроскопического) гематурия может возникнуть. По мере прогрессирования заболевания белок начинает появляться в моче. Сейчас это считается показанием для лечения ингибиторами АПФ.
Потеря слуха
Синдром Альпорта также может вызывать потерю слуха, хотя некоторые пациенты не страдают.[7] Слух у пациентов с синдромом Альпорта нормальный при рождении. Потеря слуха у пораженных пациентов прогрессирует, обычно на той стадии, когда функция почек остается нормальной, но при этом наблюдается значительная протеинурия. Однако у некоторых пациентов потеря слуха отмечается только после потери функции почек. Характерно, что для ранних изменений характерно снижение способности слышать высокочастотные звуки, «потеря слуха на высоких тонах». Это становится более серьезным и влияет также на более низкие частоты. При синдроме Альпорта потеря слуха обычно не бывает полной; хорошее общение почти всегда возможно с использованием слуховых аппаратов. [8][нужна цитата
]
Лейомиоматоз
В некоторых семьях с синдромом Альпорта сообщалось о диффузном лейомиоматозе пищевода и трахеобронхиального дерева. Симптомы обычно появляются в позднем детстве и включают дисфагию, постпрандиальную рвоту, боль в груди или эпигастрии, рецидивирующий бронхит, одышку, кашель и стридор. Лейомиоматоз подтверждается компьютерной томографией (КТ) или магнитно-резонансной томографией (МРТ).[9]
Изменения глаз
Часто наблюдаются различные глазные аномалии, в том числе: лентиконус, кератоконус, катаракта а также пятна на сетчатке пятно и средняя периферия.[10] Они редко угрожают зрению. Лентиконус (конусообразный хрусталик) можно лечить заменой хрусталика, как при катаракте. Легкий кератоконус можно лечить с помощью твердого, склерального, комбинированного или другого специального медицинского лечения. контактные линзы; прогрессирующие случаи можно остановить с помощью перекрестного связывания коллагена роговицы; и в тяжелых случаях может потребоваться трансплантат роговицы.
Другие отклонения
Расслоение аорты очень редко описывается у пациентов с ранним началом заболевания.[6] Лейомиомы, опухоли гладкой мускулатуры, поражающие пищевод и женские половые пути, могут возникать при редком синдроме перекрытия с вовлечением соседних COL4A5
и
COL4A6
гены.[11]
Синдром Альпорта — лечение
В настоящее время вылечить синдром Альпорта не представляется возможным. Терапия ограничивается борьбой с симптомами и остановкой прогрессирования заболевания. В зависимости от степени хронической почечной недостаточности — включена специальная поддерживающая терапия; а на запущенной стадии — полная замена поврежденных почек — так называемые заместительная почечная терапия, например диализ.
Если заболевание значительно прогрессирует, можно рассмотреть вопрос о трансплантации почки . Однако из-за семейной обременительности при синдроме Альпорта (возможность скрытых форм заболевания у родственников) выбор донора почки для трансплантации очень тщательный. В случае повышенных показателей артериального давления используются гипотензивные препараты.
Методы генной терапии находятся в экспериментальной фазе, что дает надежду на возможность лечения аномалий в генетическом материале, вызывающих синдром Альпорта.
Патофизиология
Генетика
Синдром Альпорта вызывается мутации в COL
4A3,
COL4A4
, и
COL4A5
, три из шести человеческих генов, участвующих в базальная мембрана (тип IV) биосинтез коллагена. Мутации в любом из этих генов препятствуют правильному производству или сборке специализированных коллаген IV типа Сеть «345», которая является важным структурным компонентом базальных мембран в почка, внутренний ухо, и глаз.[12] Он также встречается в других местах, включая альвеолы легких. Базальные мембраны — это тонкие листовые структуры, которые разделяют и поддерживают клетки во многих тканях. Коллаген типа IV «112» типа обнаружен как у позвоночных, так и у беспозвоночных, и является основной изоформой в большинстве базальных мембран человека. Когда мутации предотвращают образование коллагеновой сети 345 типа IV в клубочках, сеть 112, которая формируется в процессе внутриутробного развития, но обычно заменяется на 345, сохраняется и во взрослой жизни.[
нужна цитата
]
Шаблоны наследования
Синдром Альпорта может иметь разные модели наследования в зависимости от того, какая конкретная мутация присутствует.
- У большинства людей с синдромом Альпорта (около 85%) заболевание передается по наследству. Х-связанный шаблон,[13] из-за мутаций в COL4A5
ген. Состояние считается Х-сцепленным, если ген, участвующий в заболевании, расположен на Х хромосома. У мужчин, у которых есть только одна Х-хромосома, одна измененная копия
COL4A5
гена достаточно, чтобы вызвать тяжелый синдром Альпорта, что объясняет, почему у большинства пораженных мужчин в конечном итоге развивается почечная недостаточность. У женщин, имеющих две Х-хромосомы, мутация в одной копии
COL4A5
ген обычно приводит к образованию крови в моче, но у большинства пораженных женщин почечная недостаточность не развивается. - Синдром Альпорта также может передаваться по наследству аутосомно-рецессивный шаблон, если обе копии COL4A3
или же
COL4A4
ген, расположенный на хромосома 2, были мутированы.[12] Чаще всего родители ребенка с аутосомно-рецессивным заболеванием не страдают, но являются носителями одной копии измененного гена.[
нужна цитата
] - Предыдущие описания аутосомно-доминантный форма теперь обычно классифицируется как другие условия.[14] В частности, состояния, связанные с гигантскими тромбоцитами и связанные с мутациями MYH9
больше не считаются вариантами Alport. Однако очевидная аутосомно-доминантная передача заболевания, связанная с мутациями в
COL4A3
и
COL4A4
действительно происходит.[15][16]
Клиническая полезная генная карта для: синдрома Альпорта.[17]
Pathophysiology[edit]
Genetics
Alport syndrome is caused by mutations in COL
4A3,
COL4A4
, and
COL4A5
, three of six human genes involved in basement membrane (type IV) collagen biosynthesis. Mutations in any of these genes prevent the proper production or assembly of the specialised type IV collagen ‘345’ network which is an important structural component of basement membranes in the kidney, inner ear, and eye. It is also found in other locations, including the alveoli of the lungs. Basement membranes are thin, sheet-like structures that separate and support cells in many tissues. Type IV collagen ‘112’ type is found in both vertebrates and invertebrates, and is the major isoform in most human basement membranes. When mutations prevent the formation of 345 type IV collagen network in the glomerulus, the 112 network, which is formed in fetal development but usually replaced by 345, persists into adult life.[
citation needed
]
Inheritance patterns
Alport syndrome can have different inheritance patterns depending on which specific mutation is present.
- In most people with Alport syndrome (about 85%), the condition is inherited in an X-linked pattern, due to mutations in the COL4A5
gene. A condition is considered X-linked if the gene involved in the disorder is located on the X chromosome. In males, who have only one X chromosome, one altered copy of the
COL4A5
gene is sufficient to cause severe Alport syndrome, explaining why most affected males eventually develop kidney failure. In females, who have two X chromosomes, a mutation in one copy of the
COL4A5
gene usually results in blood in the urine, but most affected females do not develop kidney failure. - Alport syndrome can also be inherited in an autosomal recessive pattern if both copies of the COL4A3
or
COL4A4
gene, located on chromosome 2, have been mutated. Most often, the parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene.[
citation needed
] - Past descriptions of an autosomal dominant form are now usually categorized as other conditions. Notably, conditions associated with giant platelets and associated with mutations of MYH9
are no longer considered to be Alport variants. However apparent autosomal dominant transmission of disease associated with mutations in
COL4A3
and
COL4A4
does occur.
Clinical utility gene card for: Alport syndrome.
Диагностика
Диагноз обычно может быть поставлен на основании сочетания клинических критериев, критериев семейного анамнеза и биопсии.
Биопсия почек или кожи
Биопсия почек должна быть полезной до того, как болезнь станет слишком распространенной. Изменения при обычной (световой) микроскопии не характерны, и возможность других диагнозов, особенно фокально-сегментарного гломерулосклероза (ФСГС), может быть увеличена. Электронная микроскопия показывает характерную последовательность изменений от истончения базальной мембраны клубочка (GBM) до областей истончения и утолщения и, наконец, до сложного внешнего вида с очевидным расщеплением, часто описываемого как вид «плетеной корзины». Ранние или очень локализованные изменения в этом спектре не являются диагностическими, но более поздние изменения считаются диагностическими.[нужна цитата
]
Иммуногистохимические или иммунофлуоресцентные исследования для идентификации белков COL3-4-5 в GBM могут быть полезны. Однако эти исследования могут быть нормальными для некоторых пациентов с синдромом Альпорта, особенно в более легких вариантах.
Кожа содержит коллаген IV типа в сети «556». Биопсия кожи использовалась, чтобы показать отсутствие COL4A5
генный продукт, но эти методы не просты, применимы только к пациентам с тяжелой
COL4A5
мутации, и широко не доступны. Генетическое тестирование — теперь лучшая альтернатива, если биопсия почки невозможна.[
нужна цитата
]
История семьи
Семейный анамнез терминальной стадии почечной недостаточности с нарушением слуха наводит на мысль о синдроме Альпорта, но другие состояния могут вызывать эту комбинацию аномалий. Большинство из них можно отличить по клиническим признакам. Обнаружение гематурии у родственников наводит на размышления.[нужна цитата
]Хотя Х-сцепленное наследование является наиболее распространенным паттерном, генетическое тестирование показывает, что атипичные проявления могут быть более распространенными, чем считается в настоящее время.
Генетическое тестирование
Генетическое тестирование играет все более важную роль в подтверждении диагноза, когда клинические признаки не являются доказательством.[нужна цитата
]
Другие тесты
Было предложено использовать осмотр глаз для скрининга.[18]
Особенности клинической картины и лечения синдрома Альпорта у детей
Заподозрить наличие патологии у малыша можно сразу же после его появления на свет. Дети имеют характерные изменения и деформации лицевого черепа в виде расщепления верхней губы (заячья губа) и воронкообразного твёрдого нёба (волчья пасть), увеличенные размеры головы, а также типичное выражение лица с блуждающим взглядом. Другие мутации могут проявляться в форме нарушения слуха и зрения. Такой ребёнок не реагирует даже на громкие звуки и резкие движения окружающей среды, что обнаруживается ещё в родильном доме.
Для устранения внешних дефектов маленьким пациентам проводится пластическая операция. Мне довелось участвовать в ушивании верхней губы ребёнку с такой проблемой. После хирургического вмешательства остаётся небольшой шрам, который со временем бледнеет и становится практически незаметным. Операции проводятся в течение нескольких недель после рождения ребёнка, что снижает риск возникновения вторичных осложнений.
Для профилактики повреждений почечной ткани маленьким пациентам назначается специальная диета. Новорождённые и младенцы находятся на грудном вскармливании или получают максимально адаптированную к составу женского молока смесь. Это позволяет устранить риск развития иммунного дефицита. Основные средства для терапии симптоматических признаков заболевания вводятся на 5–7 году жизни (раньше — по потребности) и практически не отличаются от лечения, которое получает взрослый человек. Для адаптации слепых и глухих детей в обществе используют слуховые аппараты, корректирующие операции. Первые несколько лет ребёнок должен посещать специальные школы или дополнительно заниматься со специалистом, чтобы нагнать сверстников.
Уход
Заболевание почек и почечная недостаточность
Помимо мер по хроническая болезнь почек (ХБП) любой причины, есть доказательства того, что Ингибиторы АПФ может замедлить ухудшение функции почек при синдроме Альпорта, отсрочив необходимость диализа или трансплантации.[19] Развитие протеинурии рекомендовано как показание к началу лечения.[6]
После развития почечной недостаточности пациенты обычно хорошо себя чувствуют. диализ или с трансплантацияпочки. Трансплантация редко может быть связана с образованием антител к коллагену IV типа в донорской почке, что приводит к прогрессирующей недостаточности трансплантата в результате Синдром Гудпасчера («Посттрансплантационная болезнь Альпорта против GBM»).[20][21]
Генная терапия часто обсуждался, но доставив его подоциты в клубочках, которые обычно производят коллаген типа IV в базальной мембране клубочков, является сложной задачей.[22]
Потеря слуха
Неизвестно, влияют ли ингибиторы АПФ или другие методы лечения на потерю слуха. Людям с классическим синдромом Альпорта слуховые аппараты часто требуются в подростковом или молодом возрасте.[нужна цитата
]
Синдром Альпорта — осложнения
Симптомы нефротического синдрома, вызванные чрезмерной потерей белка поврежденными клубочками, появляются позже (примерно в возрасте 10-15 лет) и беспокоят половину пациентов. Нарушение кровотока через поврежденные почки приводит к развитию артериальной гипертензии (артериальное давление> 140/90 мм рт. Ст. У взрослых).
Хроническая почечная недостаточность со временем прогрессирует , что приводит к серьезным нарушениям функции почек (терминальная стадия почечной недостаточности). У девочек, если развиваются симптомы, это преходящая гематурия, чрезмерная потеря белка почками (протеинурия) встречается реже, болезнь протекает в легкой форме, а симптомы тяжелого поражения почек развиваются в более позднем возрасте (50-75 лет).
Рекомендации
- «Заболевания почек: синдром Альпорта». Архивировано из оригинал на 2004-06-12. Получено 2004-06-16.
- «Что такое синдром Альпорта?». Синдром Альпорта
. Получено 2019-01-06. - «Синдром Альпорта» в Медицинский словарь Дорланда
- Лагона Е., Царцали Л., Костариду С., Скиатиту А., Георгаки Е., Социу Ф. (апрель 2008 г.). «Биопсия кожи для диагностики синдрома Альпорта». Гиппократия
.
12
(2): 116–8. ЧВК 2464308. PMID 18923659. - Alport AC (март 1927 г.). «Наследственный семейный врожденный геморрагический нефрит». Британский медицинский журнал
.
1
(3454): 504–6. Дои:10.1136 / bmj.1.3454.504. JSTOR 25322864. ЧВК 2454341. PMID 20773074. - ^ абc
UK Alport Group (25 июля 2013 г.). «Информация для врача по Синдрому Альпорта».
Редкий
. Реестр редких заболеваний почек. Получено 17 февраля 2021. - Чжоу Дж., Герц Дж. М., Трюггвасон К. (июнь 1992 г.). «Мутация в цепи коллагена альфа 5 (IV) при синдроме Альпорта с ювенильным началом без потери слуха или поражения глаз: обнаружение денатурирующим градиентным гель-электрофорезом продукта ПЦР». Американский журнал генетики человека
.
50
(6): 1291–300. ЧВК 1682577. PMID 1598909. - Уотсон С., Падала С.А., Буш Дж. С. (28 мая 2021 г.). «Синдром Альпорта». StatPearls. PMID 29262041. Получено 30 мая 2021. Цитировать журнал требует | журнал = (помощь)
- Синдром Альпорта ~ клинический
в eMedicine - Чу К.С., Сакхуджа В., Агарвал А., Джа В., Джоши К., Датта Б.Н. и др. (1993). «Наследственный нефрит (синдром Альпорта) — клинический профиль и наследование в 28 родословных». Нефрология, Диализ, Трансплантация
.
8
(8): 690–5. Дои:10.1093 / ndt / 8.8.690. PMID 8414153. - Каштан СЕ (февраль 2021 г.). «Синдром Альпорта». В Adam MP, Ardinger HH, Pagon RA, et al. (ред.). Джин отзывы
. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл. - ^ аб
Нозу К., Наканиси К., Абэ Й., Удагава Т., Окада С., Окамото Т. и др. (Февраль 2019). «Обзор клинических характеристик и генетического фона синдрома Альпорта».
Клиническая и экспериментальная нефрология
.
23
(2): 158–168. Дои:10.1007 / s10157-018-1629-4. ЧВК 6510800. PMID 30128941. - Джейс Дж. П., Кнебельманн Б., Гиатрас И., Де Марчи М., Риццони Г., Реньери А. и др. (Октябрь 2003 г.). «Х-сцепленный синдром Альпорта: естественное течение жизни и корреляции генотип-фенотип у девочек и женщин, принадлежащих к 195 семьям: исследование« Совместные действия по синдрому Альпорта Европейского сообщества »». Журнал Американского общества нефрологов
.
14
(10): 2603–10. Дои:10.1097 / 01.ASN.0000090034.71205.74. PMID 14514738. - «Синдром Альпорта, аутосомно-доминантный». Онлайн-менделевское наследование в человеке (OMIM)
. Университет Джона Хопкинса. Получено 2008-11-24. - Харрат М., Макни С., Макни К., Каммун К., Чарфеддин К., Азаиз Х. и др. (Сентябрь 2006 г.). «Аутосомно-доминантный синдром Альпорта: исследование большой тунисской семьи». Саудовский журнал болезней почек и трансплантации
.
17
(3): 320–5. PMID 16970251. - Пескуччи С., Мари Ф., Лонго И., Вогиаци П., Казелли Р., Скала Е. и др. (Май 2004 г.). «Аутосомно-доминантный синдром Альпорта: естественное течение болезни, вызванной геном COL4A3 или COL4A4». Kidney International
.
65
(5): 1598–603. Дои:10.1111 / j.1523-1755.2004.00560.x. PMID 15086897. - Герц Дж. М., Томассен М., Стори Х., Флинтер Ф. (июнь 2012 г.). «Клиническая полезная генная карта для: синдрома Альпорта». Европейский журнал генетики человека
.
20
(6): 713. Дои:10.1038 / ejhg.2011.237. ЧВК 3355248. PMID 22166944. - Чжан К.В., Колвилл Д., Тан Р., Джонс К., Александр С.И., Флетчер Дж., Савиг Дж. (Август 2008 г.). «Использование глазных аномалий для диагностики X-сцепленного синдрома Альпорта у детей». Детская нефрология
.
23
(8): 1245–50. Дои:10.1007 / s00467-008-0759-4. PMID 18343956. S2CID 28650514. - Синдром Альпорта ~ лечение
в eMedicine - «Синдром Альпорта». Почечное отделение в Королевской больнице Эдинбурга, Шотландия
. - «EdRen — Edinburgh Royal Infirmary Renal Unit — болезнь Альпорта, направленная против GBM». www.edren.org
. Получено 2016-02-17. - Трюггвасон К., Хейккила П., Петтерссон Э., Тибелл А., Торнер П. (май 1997 г.). «Можно ли лечить синдром Альпорта с помощью генной терапии?». Kidney International
.
51
(5): 1493–9. Дои:10.1038 / ки.1997.205. PMID 9150464. - Temme J, Kramer A, Jager KJ, Lange K, Peters F, Müller GA, et al. (Декабрь 2012 г.). «Результаты лечения пациентов мужского пола с синдромом Альпорта, проходящих заместительную почечную терапию». Клинический журнал Американского общества нефрологов
.
7
(12): 1969–76. Дои:10.2215 / CJN.02190312. ЧВК 3513741. PMID 22997344. - Чен Д., Джефферсон Б., Харви С.Дж., Чжэн К., Гартли С.Дж., Джейкобс Р.М., Торнер П.С. (март 2003 г.). «Циклоспорин замедляет прогрессирующее заболевание почек синдрома Альпорта (Х-сцепленный наследственный нефрит): результаты на собачьей модели». Журнал Американского общества нефрологов
.
14
(3): 690–8. Дои:10.1097 / 01.ASN.0000046964.15831.16. PMID 12595505.
Эта статья включаетматериалы общественного достояния от Национальная медицинская библиотека США документ: «Синдром Альпорта». (Домашний справочник по генетике)