Введение
Туберозный склероз (ТС) — это генетически детерминированное заболевание из группы факоматозов[], отличающееся широким спектром клинических проявлений и прогрессирующим течением, которое приводит к сокращению продолжительности жизни и инвалидизации пациентов.
При этом заболевании развиваются множественные доброкачественные опухоли (гамартомы) в различных органах, включая головной мозг, глаза, кожу, сердце, почки, печень, легкие, желудочно-кишечный тракт, эндокринную и костную системы. Постепенно прогрессируя и увеличиваясь в размерах, они нарушают функции этих органов, иногда приводя к фатальным последствиям.
Данные рекомендации представляют собой протокол ведения больных ТС. Они включают критерии диагностики и вопросы профилактики ТС, в том числе пренатальную диагностику, а также современные возможности терапии этого заболевания. Данные соответствующих клинических исследований приведены с учетом уровня их доказательности.
Рекомендации предназначены для врачей-неврологов, эпилептологов, педиатров, медицинских генетиков, нейрохирургов, урологов, нефрологов, офтальмологов, организаторов здравоохранения и других специалистов, вовлеченных в диагностику и лечение больных ТС[].
Патологическая анатомия
Рис.1. Макропрепарат правого полушария головного мозга (медиальная поверхность) при туберозном склерозе: стрелками указаны множественные бугры в коре головного мозга и боковом желудочке.
Головной мозг при Туберозном склерозе обычно увеличен по сравнению с возрастной нормой, однако иногда наблюдают и микроцефалию (см.). Кора головного мозга на отдельных различной величины участках белесовато-желтоватого цвета и большей, по сравнению с окружающей тканью, плотности. В центре участков измененной коры имеются неглубокие западения, в к-рые погружаются сосуды. На разрезах полушарий головного мозга в коре обнаруживается различное количество грибовидных опухолеподобных образований, или бугров (рис. 1); выявление их легло в основу названия заболевания. Единичные или множественные сероватые бугорки размером от булавочной головки до нескольких сантиметров в диаметре также обнаруживают в белом веществе полушарий головного мозга, в боковых желудочках, чаще вблизи ленты таламуса, реже в передних и нижних рогах боковых желудочков, в сильвиевом водопроводе (водопровод среднего мозга,Т.) и в четвертом желудочке. В нек-рых случаях Т. с. изменения имеются и в мозжечке. Опухолеподобные образования могут нарушать отток цереброспинальной жидкости из желудочков и приводить к развитию внутренней гидроцефалии (см.). Эти образования обычно обызвествлены, причем очаги обызвествления расположены чаще в глубине бугра.
Рис. 2. Микропрепарат коры головного мозга при туберозном склерозе: стрелкой указан крупный атипичный нейрон; импрегнация по Бильшовскому; х 400. Рис. 3. Микропрепарат белого вещества полушария головного мозга при туберозном склерозе: 1 ~ крупная атипичная клетка; 2 — гнездное скопление клеток со сморщенной цитоплазмой и гиперхромными ядрами: окраска гематоксилин-эозином; х 400.
При гистологическом исследовании «туберозных» участков в коре головного мозга обнаруживают атипичные, причудливой формы крупные нейроны (рис. 2), аксоны к-рых нередко направляются к поверхности коры, сплетаются в клубки, внедряются в стенки сосудов. В коре обнаруживают и крупные атипичные клетки с одним-двумя пузырьковидными ядрами, цитоплазма к-рых при окрашивании тионином приобретает бледно-голубую окраску. Эти клетки, как установлено современными иммунологическими исследованиями, также являются нейронами, а не астроцита-ми, как предполагалось ранее. В коре головного мозга имеются и нарушения нормальной цитоархитектоники с отсутствием нейронов, их атрофией и вакуолизацией. Кора головного мозга нечетко отграничена от белого вещества, в ней определяются выраженный глиоз (см.), аргентофильные образования, зернистые шары, расположенные по ходу сосудов (появление зернистых шаров нек-рые исследователи связывают с деструктивными процессами, обусловленными частыми эпилептическими припадками). Стенки сосудов мозга утолщены, отмечаются признаки фиброза и гиалиноза (см.). Мягкая и паутинная оболочки головного мозга утолщены, в последней увеличено количество меланофоров. В буграх, расположенных в белом веществе полушарий головного мозга и мозжечка, обнаруживают единичные крупные атипичные клетки с пузырьковидным ядром, а также гнездные скопления клеток с гиперхромными ядрами и сморщенной цитоплазмой (рис. 3). Опухолеподобные образования в мозге больных Т.е. содержат в одних случаях крупные округлые клетки, похожие на атипичные нейроны, в других — округлые или веретенообразные клетки с овальными либо удлиненными ядрами или многоядерные клетки. Эти образования по гистол. строению сходны с невриномами (см.), субэпендимарными астроцитомами (см.), эпендимомами (см.). Узлы, обнаруживаемые в глазном яблоке, чаще всего являются глиомами. Описаны также очаговые обызвествления в сосудистой оболочке глазного яблока, ее ангиомы (см.), пороки развития глаза.
В миокарде больных Т. с. также обнаруживают опухолеподобные узлы, нередко расположенные в области проводящей системы сердца или растущие в полости сердца и имеющие строение липомы (см.), фибролипомы, рабдомиомы (см.). Известны наблюдения рабдомиоматоза миокарда (скопления атипичных клеток в миокарде). В почках при Т. с. обнаруживают мелкие и гигантские гамартомы (см.), капиллярные ангиомы, а также другие пороки развития сосудов и различных отделов нефрона. Описаны также липомиомы и «саркоматозная ткань» в лимфатических узлах, липомиомы, ангиомиолипомы (см. Почки) и ангиоматозные аденомы (см. Ангиоматоз) в печени и селезенке, кисты и миоматозные образования в легких, скопления жировых и атипичных эпителиальных клеток в щитовидной железе, аплазия и гипоплазия яичек, липоматозные образования в надпочечниках, аденомы (см.) из клеток панкреатических островков, скопления клеток многослойного плоского эпителия в передней доле гипофиза, кисты в задней его доле и др.
Классификация, этиология и патогенез ТС
Выделяют ТС 1-го типа, обусловленный мутацией гена TSC1
, и ТС 2-го типа, обусловленный мутацией гена
TSC2
. Считается, что у пациентов с мутацией TSC1 заболевание течет более мягко, а мутации в гене
TSC2
обусловливают более тяжелое развитие патологии.
Факторы риска отсутствуют, так как заболевание генетическое (моногенное).
Частота ТС в популяции составляет 1:10 000 (у новорожденных — 1:6000). Расчетное число больных ТС в Российской Федерации около 7000 человек, поэтому ТС относится к редким (орфанные) заболеваниям.
ТС — аутосомно-доминантное генетически гетерогенное заболевание с неполной пенетрантностью, вариабельной экспрессивностью и высокой частотой возникновения новых (спонтанных) мутаций, которые обнаруживаются в 68% всех случаев, дебютирующих в раннем возрасте. Приблизительно от 10 до 30% случаев ТС обусловлено мутациями в гене TSC1 (OMIM 605284) (ТС 1-го типа, OMIM #191100), локализованном на 9 хромосоме в районе 9q34, который кодирует белок гамартин. Остальные случаи болезни обусловлены мутациями в гене TSC2 (OMIM 191092) (ТС 2-го типа — OMIM #613254), локализованном на 16 хромосоме в районе 16p13 и кодирующим белок туберин. Для генов ТС характерны высокая пенетрантность (до 100%) и вариабельная экспрессивность, прослеживающаяся при семейных случаях заболевания, когда у родственников с одной и той же семейной доминантной мутацией может различаться тяжесть заболевания.
Гены TSC1
и
TSC2
в норме являются естественными генами-супрессорами опухолевого роста. Белковые продукты генов
TSC1
и
TSC2
, гамартин и туберин, образуют гетеродимер, способный ингибировать опосредованный комплексом mTORС1 (mammalian Target of Rapamycin Complex 1) сигнальный каскад. Механизм патогенеза ТС состоит в мутациях в генах
ТCS1
и
TSC2
с потерей их функции и связанной с мутациями патологической активацией киназы mTOR. В результате происходит активация пути сигнальной передачи PI3K/Akt/mTOR. Данный каскад является ключевым регулятором роста и пролиферации клеток. Он активируется в ответ на поступление питательных веществ и факторов роста, регулируя ряд клеточных функций, таких как трансляция, транскрипция и аутофагия. Гиперактивация каскада mTORС1, ведущая к усилению клеточной пролиферации, считается важным звеном злокачественной трансформации. В клетках с мутацией
TSC1
и
TSC2
данный сигнальный путь постоянно «включен». Этот путь сигнальной трансдукции является ключевым звеном патогенеза ТС. Одна из мутаций
TSC1
и
TSC2
содержится во всех клетках организма.
В клетке-родоначальнице опухолевого клона происходит инактивация второго, незатронутого наследственной мутацией, аллеля.
Как проявляется туберозный склероз?
Симптоматика болезни Бурневилля-Прингла представлена разными типами проявлений. Она состоит из:
- Расстройства ЦНС.
- Дерматологических изменений.
- Офтальмологических нарушений.
- Образования опухолей.
Начало развития аномалии происходит в разные временные отрезки. Зачастую, врачи диагностируют ее первые пять лет жизни ребенка. Существуют вариабельные течения заболевания. Встречаются легкие случаи отклонения, которые проявляются факультативными симптомами неспецифической формы. Стертый тип ТС протекает без наличия припадков эпилепсии, низкого уровня интеллекта и сбое в поведении.
Нарушения ЦНС
Проблемы с нервной системой занимают главенствующее место. В более 80% ситуациях у пациентов диагностируют судорожный синдром, которым начинается первый этап развития аномалии. У больных выделяют такие проблемы:
- Спазмы инфантильной природы, перерастающие в синдром Леннокса-Гасто.
- Пароксизмы соматического и сенсомоторного характера, вызывающие задержку в психическом развитии. Вызывают высокий уровень агрессии, аутизм, СДВГ.
- Атипичная эпилепсия.
- Низкая степень интеллектуальной способности.
- Постоянное беспокойство.
- Капризность и недовольство.
- Заторможенность и трудности в концентрации внимания.
- Проблемы со сном.
Деформации дерматологического характера
Кожные высыпания встречаются у 100% пациентов. Обычно, врачи наблюдают появление светлых пятен (гипопигментация), распространяющихся в промежутке трех лет после рождения ребенка. В ходе взросления они только увеличивают свое число. Локализуются в любых местах на теле. Дополнительно образуются:
- Ангиофибромы — представлены большим скоплением или одиночными единицами плотных узлов в форме зерна. Обладают красным или желтым цветом.
- «Шагреневая кожа» — неодинаковые зоны огрубевшего эпидермиса, которые развиваются сзади (спина, поясница). Формируется в момент с 10 до 20 лет. Размеры составляют от 2 до 10 сантиметров.
- Бляшки фиброзного происхождения — встречается в 25% случаях.
- Дерматофиброз — наблюдается у 30% больных.
Офтальмологические проблемы
Достаточно редкое явление, но иногда врачи отмечают признаки нарушения у половины пациентов с ТС — присутствие доброкачественных новообразований в районе зрительного нерва или на сетчатки. Гамартомы представлены полоской формой с гладкой поверхностью, которая может возвышаться или быть узловатой. Их главной особенностью считается влияние на снижение уровня зрения. Дополнительные сбои офтальмологического аппарата:
- Процесс депигментация радужки.
- Образование отечности диска.
- Отсутствие части оболочки.
- Косоглазие.
- Катаракта.
- Появление ангиофибромы на веках.
Сбои в функционировании внутренних органов
Опухоли, которые сопровождают ТС выделяются большим количеством и частыми поражениями двустороннего типа парных органов тела. Они незаметно протекают в начале развития заболевания. Период проявления имеет широкий диапазон — от 5 до 40 лет. У больных может сформироваться:
- Рабдомиома сердца.
- Кисты в легочном аппарате.
- Поликистоз в почечном отделе.
- Гамартомы в области печени.
- Полипы реклатольного происхождения.
Существует вероятность прогрессирования злокачественных новообразований.
Нарушения сердечно-сосудистого аппарата
В этом районе согласно медицинским документам в более 30% случаях врачи выявляют опухоли (рабдомиомы) главного двигателя тела. Во время вынашивания ребенка может произойти выкидыш, вызванный антенатальной природой. В детском возрасте врачи наблюдают:
- Возникновение аритмии.
- Синдрома WPW.
- Появление тахикардии.
- Фибрилляция желудочков.
Встречается поражение легочного аппарата (киста) у пациентов после 30 лет. Нарушения в функционировании ЖКТ представлено появлением новообразований в полости рта, изменениями поверхностного слоя покрытия зубов, прогрессированием гамартомы в области печени. Аномальные реакции в почках занимают второе место после проблем с нервной системой, которые способны вызвать смерть.
Дифференциальная диагностика
Диагностическое отграничение ТС проводится в основном по отношению к другим нейро-кожным синдромам, при которых также наблюдаются пятна депигментации (табл. 3)
.
Важно отметить, что при подозрении на ТС пациент должен быть обследован у специалистов различного профиля для исключения или подтверждения патологии внутренних органов. Такая мультидисциплинарная команда должна состоять в пренатальном периоде из генетика, акушера-гинеколога и кардиолога, в возрасте от 0 до 1 года — из детского невролога и дерматолога, в возрасте 1-5 лет — дополняться офтальмологом, в возрасте 5-18 лет — должны привлекаться нефролог и уролог, после 18 лет — пульмонолог.
Профилактика
В мировой практике профилактика заболевания сводится к пренатальной диагностике ТС. При спорадическом ТС риск повторного рождения больного ребенка составляет 2%, при наследственном — 50%. Поэтому при подозрении на наследственный характер ТС (при наличии подозрения или подтвержденного диагноза ТС у будущей матери, отца или родственников) генетическую диагностику ТС необходимо проводить еще при планировании беременности. Подтверженный наследственный тип ТС является основанием для проведения инвазивной пренатальной диагностики. Также при подозрении на ТС у плода рекомендуется проведение ЭхоКГ на сроках 20-24 нед беременности для исключения рабдомиомы сердца.
В Российской Федерации этот метод профилактики не получил еще должного распространения. В настоящий момент мы можем профилактировать развитие только жизнеугрожающих осложнений со стороны ЦНС и почек.
Существуют зарубежные исследования IV уровня доказательности, свидетельствующие о том, что назначение антиэпилептической терапии пациентам 1-го года жизни, еще не страдающим эпилепсией, но уже имеющим эпилептиформные разряды на ЭЭГ, приводит к тому, что эпилепсия не развивается, и дети в дальнейшем не страдают умственной отсталостью.
Терапия
Этиологическое лечение при ТС отсутствует. До 2012 г. лечение носило симптоматический характер. В 2012 г. был зарегистрирован препарат эверолимус (торговое название — афинитор), который влияет на основное звено патогенеза при ТС (является ингибитором сигнального пути mTOR) и уменьшает рост опухолей в ЦНС и почках (уровень доказательности I) [29]. Препарат эверолимус в форме таблеток включен в Перечень жизненно необходимых и важнейших лекарственных средств (ЖНВЛС)[].
Имеются отдельные работы, указывающие на то, что он также уменьшает число эпилептических приступов (уровень доказательности IV) и снижает степень выраженности ангиофибром лица (уровень доказательности I).
Далее рассматриваются отдельные аспекты лечения в зависимости от симптомов и их степени выраженности.
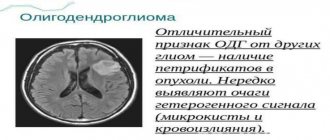
Лечение СЭГА
При наличии СЭГА с клиническими проявлениями показано оперативное лечение — удаление опухоли. Не выявленная своевременно внутричерепная гипертензия, обусловленная СЭГА, является наиболее частой причиной смерти у 50% больных ТС старше 10 лет. Редкая причина смерти — кровоизлияние в опухоль.
Обоснованием для более раннего хирургического удаления опухоли являются: 1) рост опухоли и появление клинических симптомов после предшествующего бессимптомного течения СЭГА ввиду локализации вблизи отверстия Монро, сопровождающийся развитием обструкции, нарушениями ликвородинамики, внутрижелудочковым кровотечением; 2) рост СЭГА, сопровождающийся ухудшением течения эпилепсии, что ассоциировано с развитием гидроцефалии и/или прямым механическим раздражением межжелудочковой перегородки; 3) СЭГА большого размера, которые могут деформировать отверстие Монро, что затрудняет удаление опухоли и гемостаз во время операции в непосредственной близости других структур (свод, каудальные ядра, эпендима, вены эпендимы, межжелудочковая перегородка); 4) в большинстве случаев осложнения в послеоперационном периоде развиваются при выполнении операции детям с симптомным течением СЭГА и признаками повышения внутричерепного давления или гидроцефалией до операции [25].
Диагноз
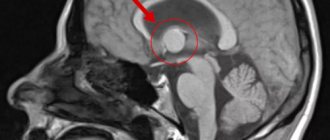
Диагноз основывается на появлении эпилептических припадков полиморфного характера в раннем детском возрасте, отставании умственного развития, задержке развития моторных и речевых функций, наличии аденом сальных желез на лице и других характерных изменений кожи, изменении на глазном дне, прогрессировании болезни, семейном характере заболевания. При рентгенографии черепа обнаруживают остеосклероз (см.) преимущественно свода черепа, признаки внутричерепной гипертензии и петрификаты (см.) в структурах мозга, к-рые по виду напоминают очаги обызвествления при токсоплазмозе и цитомегалии. Правильной и ранней диагностике помогают данные компьютерной томографии (см. Томография компьютерная), обнаруживающей изменения системного характера в головном мозге, почках, печени, сердце и др. При компьютерной томографии головного мозга выявляют множественные опухолевидные образования, расширение желудочков, гидроцефалию различной выраженности, петрификаты, к-рые локализуются в коре, чаще в лобной и теменной долях, в третьем и боковых желудочках. Рентгенол. исследование позвоночника, костей таза, конечностей выявляет очаги остеосклероза, утолщение периоста, кистоподобные образования в периосте костей рук и ног, остеопороз (см.) пястных и плюсневых костей и др.
При электрофизиологическом исследовании обнаруживают фокальные или диффузные изменения ЭЭГ эпилептоидного характера (см. Электроэнцефалография). Цереброспинальная жидкость обычно не изменена, за исключением случаев, когда при тяжелых поражениях головного мозга развиваются гидроцефалия, отек мозга, эпилептический статус и другие осложняющие течение болезни состояния.
Дифференциальный диагноз
проводят с нейрофиброматозом (см.), энцефалотригеминалъным ангиоматозом (см.) болезнью Гиппеля — Линдау (см. Гиппеля — Линдау болезнь), синдромом Клиппеля — Треноне (см. Кровеносные сосуды, пороки развития), синдромом Олбрайта (см. Псевдогипопаратиреоз), синдромом Ларсена и пороками развития ц. н. с.