Introduction
Tuberous sclerosis (TS) is a genetically determined disease from the group of phakomatoses[], characterized by a wide range of clinical manifestations and a progressive course, which leads to a reduction in life expectancy and disability of patients.
In this disease, multiple benign tumors (hamartomas) develop in various organs, including the brain, eyes, skin, heart, kidneys, liver, lungs, gastrointestinal tract, endocrine and skeletal systems. Gradually progressing and increasing in size, they disrupt the functions of these organs, sometimes leading to fatal consequences.
These recommendations represent a protocol for the management of patients with TS. They include diagnostic criteria and issues of prevention of TS, including prenatal diagnosis, as well as modern treatment options for this disease. Data from relevant clinical studies are presented based on their level of evidence.
The recommendations are intended for neurologists, epileptologists, pediatricians, medical geneticists, neurosurgeons, urologists, nephrologists, ophthalmologists, health care managers and other specialists involved in the diagnosis and treatment of patients with TS [].
Pathological anatomy
Fig.1.
Macroscopic specimen of the right hemisphere of the brain (medial surface) in tuberous sclerosis: arrows indicate multiple tubercles in the cerebral cortex and lateral ventricle. The brain in tuberous sclerosis is usually enlarged compared to the age norm, but microcephaly is sometimes observed (see). The cerebral cortex in individual areas of varying sizes is whitish-yellowish in color and has a greater density than the surrounding tissue. In the center of the areas of the altered cortex there are shallow depressions into which the vessels are immersed. On sections of the cerebral hemispheres, various numbers of mushroom-shaped tumor-like formations, or tubercles, are found in the cortex (Fig. 1); Their identification formed the basis for the name of the disease. Single or multiple grayish tubercles ranging in size from a pinhead to several centimeters in diameter are also found in the white matter of the cerebral hemispheres, in the lateral ventricles, more often near the thalamic ribbon, less often in the anterior and inferior horns of the lateral ventricles, in the aqueduct of Sylvius (aqueduct of the midbrain, T .) and in the fourth ventricle. In some cases, T. s. There are also changes in the cerebellum. Tumor-like formations can disrupt the outflow of cerebrospinal fluid from the ventricles and lead to the development of internal hydrocephalus (see). These formations are usually calcified, with foci of calcification located more often in the depths of the tubercle.
Rice. 2. Microscopic specimen of the cerebral cortex in tuberous sclerosis: the arrow indicates a large atypical neuron; impregnation according to Bielschowsky; x 400. Fig. 3. Microscopic specimen of the white matter of the cerebral hemisphere in tuberous sclerosis: 1 ~ large atypical cell; 2 — nested accumulation of cells with wrinkled cytoplasm and hyperchromic nuclei: hematoxylin-eosin staining; x 400.
Histological examination of “tuberous” areas in the cerebral cortex reveals atypical, bizarrely shaped large neurons (Fig. 2), the axons of which are often directed to the surface of the cortex, intertwined into balls, and embedded in the walls of blood vessels. Large atypical cells with one or two bubble-like nuclei are also found in the cortex, the cytoplasm of which, when stained with thionin, acquires a pale blue color. These cells, as established by modern immunological studies, are also neurons, and not astrocytes, as previously assumed. In the cerebral cortex there are also disturbances of normal cytoarchitecture with the absence of neurons, their atrophy and vacuolization. The cerebral cortex is not clearly demarcated from the white matter, it contains pronounced gliosis (see), argentophilic formations, granular balls located along the vessels (some researchers associate the appearance of granular balls with destructive processes caused by frequent epileptic seizures). The walls of the brain vessels are thickened, there are signs of fibrosis and hyalinosis (see). The soft and arachnoid membranes of the brain are thickened, and the number of melanophores in the latter is increased. In the tubercles located in the white matter of the cerebral hemispheres and cerebellum, single large atypical cells with a vesicular nucleus are found, as well as nested clusters of cells with hyperchromatic nuclei and wrinkled cytoplasm (Fig. 3). Tumor-like formations in the brain of patients I.e. In some cases they contain large round cells, similar to atypical neurons, in others - round or spindle-shaped cells with oval or elongated nuclei or multinucleated cells. These formations according to gistol. similar in structure to neuromas (see), subependymal astrocytomas (see), ependymomas (see). Nodules found in the eyeball are most often gliomas. Focal calcifications in the choroid of the eyeball, its angiomas (see), and malformations of the eye are also described.
In the myocardium of patients with T. s. tumor-like nodes are also found, often located in the region of the conduction system of the heart or growing in the cavity of the heart and having the structure of lipomas (see), fibrolipomas, rhabdomyomas (see). There are known observations of myocardial rhabdomyomatosis (accumulations of atypical cells in the myocardium). In the kidneys with T. s. detect small and giant hamartomas (see), capillary angiomas, as well as other malformations of blood vessels and various parts of the nephron. Also described are lipomyomas and “sarcomatous tissue” in the lymph nodes, lipomyomas, angiomyolipomas (see Kidneys) and angiomatous adenomas (see Angiomatosis) in the liver and spleen, cysts and myomatous formations in the lungs, accumulations of fatty and atypical epithelial cells in the thyroid gland, aplasia and hypoplasia of the testicles, lipomatous formations in the adrenal glands, adenomas (see) from the cells of the pancreatic islets, accumulations of stratified squamous epithelial cells in the anterior lobe of the pituitary gland, cysts in its posterior lobe, etc.
Classification, etiology and pathogenesis of TS
Type 1 TS is distinguished, caused by a mutation of the TSC1
, and type 2 TS, caused by a mutation in the
TSC2
.
It is believed that patients with a TSC1 mutation have a milder course of the disease, while mutations in the TSC2
cause a more severe development of the pathology.
There are no risk factors, since the disease is genetic (monogenic).
The frequency of TS in the population is 1:10,000 (in newborns - 1:6000). The estimated number of patients with TS in the Russian Federation is about 7,000 people, so TS is classified as a rare (orphan) disease.
TS is an autosomal dominant genetically heterogeneous disease with incomplete penetrance, variable expressivity and a high incidence of new (spontaneous) mutations, which are found in 68% of all cases that appear at an early age. Approximately 10 to 30% of TS cases are caused by mutations in the TSC1 gene (OMIM 605284) (TS type 1, OMIM #191100), located on chromosome 9 in the 9q34 region, which encodes the hamartin protein. The remaining cases of the disease are caused by mutations in the TSC2 gene (OMIM 191092) (TC type 2 - OMIM #613254), localized on chromosome 16 in the 16p13 region and encoding the tuberin protein. TC genes are characterized by high penetrance (up to 100%) and variable expressivity, which can be observed in familial cases of the disease, when relatives with the same family dominant mutation may differ in the severity of the disease.
TSC1 genes
and
TSC2
are normally natural tumor suppressor genes.
The protein products of the TSC1
and
TSC2
, hamartin and tuberin, form a heterodimer capable of inhibiting the mTORС1 (mammalian Target of Rapamycin Complex 1) signaling cascade mediated.
The mechanism of TS pathogenesis consists of mutations in the TCS1
and
TSC2
with loss of their function and pathological activation of mTOR kinase associated with mutations.
As a result, the PI3K/Akt/mTOR signaling pathway is activated. This cascade is a key regulator of cell growth and proliferation. It is activated in response to nutrients and growth factors, regulating a number of cellular functions such as translation, transcription and autophagy. Hyperactivation of the mTORC1 cascade, leading to increased cell proliferation, is considered an important link in malignant transformation. TSC1
and
TSC2
mutations, this signaling pathway is constantly “on.”
This signal transduction pathway is a key element in the pathogenesis of TS. One of the TSC1
and
TSC2
is found in all cells of the body.
In the parent cell of the tumor clone, the second allele, unaffected by the hereditary mutation, is inactivated.
How does tuberous sclerosis manifest?
The symptoms of Bourneville-Pringle disease are represented by different types of manifestations. It consists of:
- CNS disorders.
- Dermatological changes.
- Ophthalmological disorders.
- Tumor formation.
The onset of anomaly development occurs at different time periods. Often, doctors diagnose it during the first five years of a child’s life. There are variable courses of the disease. There are mild cases of deviation, which are manifested by optional symptoms of a nonspecific form. The erased type of TS occurs without the presence of epileptic seizures, low level of intelligence and behavioral disturbances.
Central nervous system disorders
Problems with the nervous system occupy a dominant place. In more than 80% of situations, patients are diagnosed with convulsive syndrome, which begins the first stage of development of the anomaly. Patients have the following problems:
- Spasms of an infantile nature developing into Lennox-Gastaut syndrome.
- Paroxysms of a somatic and sensorimotor nature, causing a delay in mental development. Causes high levels of aggression, autism, ADHD.
- Atypical epilepsy.
- Low degree of intellectual ability.
- Constant anxiety.
- Moodiness and dissatisfaction.
- Lethargy and difficulty concentrating.
- Sleep problems.
Deformations of a dermatological nature
Skin rashes occur in 100% of patients. Usually, doctors observe the appearance of light spots (hypopigmentation) that spread within three years after the birth of the child. As they grow older, they only increase in number. Localized in any place on the body. Additionally formed:
- Angiofibromas are represented by a large cluster or single units of dense nodes in the form of a grain. They are red or yellow in color.
- “Shagreen skin” - unequal zones of roughened epidermis that develop at the back (back, lower back). Formed between the ages of 10 and 20. Dimensions range from 2 to 10 centimeters.
- Plaques of fibrous origin - occurs in 25% of cases.
- Dermatofibrosis - observed in 30% of patients.
Ophthalmological problems
A fairly rare occurrence, but sometimes doctors note signs of a disorder in half of patients with TS - the presence of benign neoplasms in the area of the optic nerve or on the retina. Hamartomas are strip-shaped with a smooth surface, which can be raised or nodular. Their main feature is considered to be the effect on reducing the level of vision. Additional failures of the ophthalmic apparatus:
- The process of depigmentation of the iris.
- Formation of disc swelling.
- Missing part of the shell.
- Strabismus.
- Cataract.
- The appearance of angiofibroma on the eyelids.
Malfunctions of internal organs
Tumors that accompany TS are distinguished by a large number and frequent lesions of the bilateral type of paired organs of the body. They occur unnoticed at the beginning of the development of the disease. The period of manifestation has a wide range - from 5 to 40 years. Patients may develop:
- Rhabdomyoma of the heart.
- Cysts in the pulmonary apparatus.
- Polycystic kidney disease.
- Hamartomas in the liver area.
- Polyps of rectal origin.
There is a possibility of progression of malignant neoplasms.
Cardiovascular disorders
In this area, according to medical records, in more than 30% of cases, doctors identify tumors (rhabdomyomas) of the main engine of the body. During pregnancy, a miscarriage may occur due to antenatal causes. In childhood, doctors observe:
- The occurrence of arrhythmia.
- WPW syndrome.
- The appearance of tachycardia.
- Ventricular fibrillation.
Damage to the pulmonary apparatus (cyst) occurs in patients over 30 years of age. Disturbances in the functioning of the gastrointestinal tract are represented by the appearance of neoplasms in the oral cavity, changes in the surface layer of the teeth, and the progression of hamartoma in the liver area. Abnormal reactions in the kidneys are second only to problems with the nervous system, which can cause death.
Differential diagnosis
Diagnostic delimitation of TS is carried out mainly in relation to other neurocutaneous syndromes, in which spots of depigmentation are also observed (Table 3)
.
It is important to note that if TS is suspected, the patient should be examined by specialists in various fields to exclude or confirm pathology of internal organs. Such a multidisciplinary team should consist in the prenatal period of a geneticist, obstetrician-gynecologist and cardiologist, at the age of 0 to 1 year - from a pediatric neurologist and dermatologist, at the age of 1-5 years - supplemented by an ophthalmologist, at the age of 5-18 years - should be involved nephrologist and urologist, after 18 years - pulmonologist.
Prevention
In world practice, disease prevention comes down to prenatal diagnosis of TS. With sporadic TS, the risk of re-birth of a sick child is 2%, with hereditary TS - 50%. Therefore, if the hereditary nature of TS is suspected (if there is a suspected or confirmed diagnosis of TS in the expectant mother, father or relatives), genetic diagnosis of TS must be carried out even when planning pregnancy. The confirmed hereditary type of TS is the basis for invasive prenatal diagnosis. Also, if TS is suspected in the fetus, it is recommended to conduct an echocardiography at 20-24 weeks of pregnancy to exclude cardiac rhabdomyoma.
In the Russian Federation, this method of prevention has not yet received proper distribution. At the moment, we can only prevent the development of life-threatening complications from the central nervous system and kidneys.
There are foreign studies of level IV evidence indicating that the prescription of antiepileptic therapy to patients of the 1st year of life who do not yet suffer from epilepsy, but already have epileptiform discharges on the EEG, leads to the fact that epilepsy does not develop, and children do not suffer in the future mental retardation.
Therapy
There is no etiological treatment for TS. Until 2012, treatment was symptomatic. In 2012, the drug everolimus (trade name Afinitor) was registered, which affects the main link in the pathogenesis of TS (it is an inhibitor of the mTOR signaling pathway) and reduces the growth of tumors in the central nervous system and kidneys (level of evidence I) [29]. The drug everolimus in tablet form is included in the List of Vital and Essential Medicines (VED)[].
There is some evidence that it also reduces the number of epileptic seizures (level IV evidence) and reduces the severity of facial angiofibromas (level I evidence).
The following discusses individual aspects of treatment depending on the symptoms and their severity.
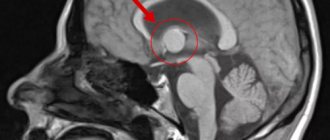
Treatment of SEGA
In the presence of SEGA with clinical manifestations, surgical treatment—tumor removal—is indicated. Untimely identified intracranial hypertension caused by SEGA is the most common cause of death in 50% of patients with TS over 10 years of age. A rare cause of death is hemorrhage into the tumor.
The rationale for earlier surgical removal of the tumor is: 1) tumor growth and the appearance of clinical symptoms after a previous asymptomatic course of SEGA due to localization near the foramen of Monro, accompanied by the development of obstruction, disturbances in liquor dynamics, and intraventricular bleeding; 2) an increase in SEGA, accompanied by a worsening of the course of epilepsy, which is associated with the development of hydrocephalus and/or direct mechanical irritation of the interventricular septum; 3) large-sized SEGAs, which can deform the foramen of Monroe, making it difficult to remove the tumor and hemostasis during surgery in the immediate vicinity of other structures (fornix, caudal nuclei, ependyma, ependymal veins, interventricular septum); 4) in most cases, complications in the postoperative period develop when surgery is performed on children with symptomatic SEGA and signs of increased intracranial pressure or hydrocephalus before surgery [25].
Diagnosis
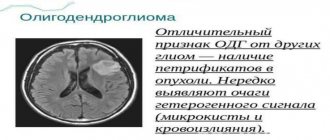
The diagnosis is based on the appearance of epileptic seizures of a polymorphic nature in early childhood, mental retardation, delayed development of motor and speech functions, the presence of adenomas of the sebaceous glands on the face and other characteristic skin changes, changes in the fundus of the eye, progression of the disease, and the familial nature of the disease. When radiography of the skull reveals osteosclerosis (see) mainly of the cranial vault, signs of intracranial hypertension and petrification (see) in the structures of the brain, which in appearance resemble foci of calcification in toxoplasmosis and cytomegaly. Correct and early diagnosis is helped by computed tomography data (see Computer Tomography), which detects systemic changes in the brain, kidneys, liver, heart, etc. Computed tomography of the brain reveals multiple tumor-like formations, dilation of the ventricles, hydrocephalus of varying severity, petrification, which are localized in the cortex, most often in the frontal and parietal lobes, in the third and lateral ventricles. X-ray examination of the spine, pelvic bones, and limbs reveals foci of osteosclerosis, thickening of the periosteum, cyst-like formations in the periosteum of the bones of the arms and legs, osteoporosis (see) metacarpal and metatarsal bones, etc.
An electrophysiological study reveals focal or diffuse EEG changes of an epileptoid nature (see Electroencephalography). Cerebrospinal fluid is usually not changed, except in cases where hydrocephalus, cerebral edema, status epilepticus and other conditions complicating the course of the disease develop in severe brain lesions.
Differential diagnosis
carried out with neurofibromatosis (see), encephalotrigeminal angiomatosis (see), Hippel-Lindau disease (see Hippel-Lindau disease), Klippel-Trenaunay syndrome (see Blood vessels, malformations), Albright syndrome (see Pseudohypoparathyroidism), syndrome Larsen and malformations of c. n. With.