Benign epithelial tumors of dysembryogenetic origin, located in the chiasmal-sellar region and in the region of the third ventricle.
Craniopharyngiomas account for 1.2-4.6% of all intracranial tumors, the incidence rate in the population is approximately 0.5-2.5 per 1,000,000 people per year. This tumor is the most common non-epileptogenic intracerebral neoplasm in children, accounting for 5 to 10% of all tumors in this age group.
According to the most widespread Erdheim theory, craniopharyngiomas develop from the remnants of the embryonic epithelium of Rathke's pouch, which represents a protrusion of the primary oral tube, from which the anterior lobe of the pituitary gland and its tuberal part develop at an early stage of the embryonic period.
Topography
Craniopharyngiomas can be divided into three topographic groups
1. Endosellar
They develop from the remnants of the epithelium preserved at the level of the pituitary gland. By the time of diagnosis, the tumor has already reached a significant size, while stretching or breaking through the diaphragm of the sella turcica and spreading suprasellar.
2. Stem
They develop from epithelial thickenings at the level of the pituitary gland stalk. They are located above the sella turcica, shifting the sella diaphragm down. The bottom of the third ventricle and the chiasm are pushed upward. The tumor capsule is separated from the third ventricle by the pia mater. Due to their topographical features, they can be considered suprasellar-extraventricular craniopharyngiomas.
In relatively rare cases, epithelial primordia can be located outside the stem, extraaxially in the subarachnoid space. Typically, these are cystic tumors of gigantic size, tending to spread throughout the subarachnoid space of the base of the brain and into the anterior, middle, and posterior cranial fossae. The tumor capsule grows “muff-like” over the optic nerves, chiasm, carotid arteries and their branches. pituitary stalk. With pronounced para- and retrosellar growth, the III, IV, V, VI, VII, VIII pairs of cranial nerves can be included in the tumor capsule.
3. Craniopharyngiomas of the third ventricle
These tumors are histogenetically associated with accumulations of embryonic epithelium in the infundibulum area. In these cases, the tumor is partially or completely located in the cavity of the third ventricle.
It is advisable to distinguish 2 topographic options: with a predominant location of the tumor in the ventricular cavity and with intra- or extraventricular localization.
Pathological anatomy
There are two main types of tumors: those consisting of dense tissue and cystic.
Rice. 6. Large craniopharyngioma cyst in the area of the sella turcica (the cerebral hemispheres are tilted posteriorly).
Craniopharyngioma is a tumor (usually in the form of a single dense node), growing slowly and expansively. Its size is from 2 to 5 cm in diameter, rarely larger (color fig. 6). Cystically degenerated areas of the tumor contain from 10 to 50 ml (in rare cases up to 200 ml) of thick liquid of yellow, amber or coffee color. K. usually has a dense capsule, quite closely connected with the surrounding brain tissue, membranes and vascular network. The tumor is supplied with blood from the branches of the arterial circle of the cerebrum.
K.'s structure can undergo significant changes over time. In its compact layers, liquefaction necrosis occurs with the formation of cysts containing fluid with a large amount of protein (from 20 to 100 ppm or more), amorphous remains of dead cellular elements and blood, cholesterol crystals, fatty acids with the deposition of salts on the inner surface of the capsule and in the tissue of the tumor itself. Histologically, cells consist of epithelial cells of varying degrees of differentiation. Along with epithelial cells of the embryonic type, epidermal type epithelium is also found. Cellular accumulations in some cases may resemble adamantinomas. In K., dystrophic changes of varying severity are always observed in the form of cyst formation, calcification, or even ossification of the stroma. K.'s capsule consists of connective or glial tissue. The diversity in the structure of the cell is regarded by a number of authors as a result of the phasic nature of its development.
Histological picture
1. Adamantinoma-like (about 85% of all craniopharyngiomas) - more common in patients of childhood and adolescence. They differ in polymorphism and can have a compact (10%), cystic (30%) and mixed (60%) structure.
2. Papillomatous (15% of all craniopharyngiomas) - unlike adamantine-like ones, this pathology is diagnosed more often in people in adulthood, usually at 30-40 years of age. These tumors are mostly solid, with little or no petrification
Clinical picture
It is characterized by a combination of the following main symptoms: endocrine metabolic disorders, decreased vision and intracranial hypertension syndrome.
Endocrine disorders are represented by an imbalance of gonadotropic hormone, luteinizing hormone, adenocorticotropic hormone (ACTH), somatotropic hormone (GH), which leads to secondary hypogonadism, hypothyroidism, hypocortisolism, diabetes insipidus, and lipid metabolism disorders.
Symptoms of hypothalamic-pituitary dysfunction:
- growth retardation and/or decreased growth rate (up to 4 cm per year) in pre-pubescent children;
-thirst, polyuria, enuresis;
- rapid increase in body weight;
-muscle weakness, fatigue;
-delayed sexual development in children;
- decreased libido and impotence in men, menstrual cycle disorders in women.
The severity of endocrine insufficiency is largely determined by the location, size of the tumor and the age of the patient.
Visual disturbances are among the early and most significant manifestations of crinopharyngiomas and in more than half of the cases are the first symptoms of the disease.
The syndrome of increased intracranial pressure is most typical for craniopharyngiomas located in the third ventricle and is caused by compression of the interventricular foramina and, less commonly, by occlusion at the level of the cerebral aqueduct.
Clinical manifestations of craniopharyngiomas may include symptoms from the midbrain, mediobasal regions of the frontal and temporal lobes, and, with pronounced retrosellar growth, signs of impact on the structures of the posterior cranial fossa.
Craniopharyngiomas
Craniopharyngiomas (CP) account for 2.5-4% of all brain tumors; Moreover, about 50% of cases occur in childhood. Peak occurrence: age 5-10 years. They arise from the anterior superior edge of the pituitary gland; they are lined with stratified squamous epithelium. Some CFs may occur primarily in the third ventricle. Almost all CFs have dense and cystic components; The fluid in cysts varies, but usually contains cholesterol crystals. CFs are benign tumors and do not undergo malignant transformation. However, the difficulty in removing them makes their course prone to frequent relapses and the need for regular monitoring and repeated surgical and radiation interventions.
What clinical manifestations are characteristic of craniopharyngiomas?
Due to the involvement of the hypothalamic-pituitary system in the pathological process, patients develop a wide variety of disorders - diabetes insipidus, hypopituitarism with typical growth retardation (in children), visual impairment (due to a primary effect on the visual pathways and secondary to intracranial hypertension), hydrocephalus . Localization is a determining factor in the difficulty of radical tumor removal, the high frequency of relapses and the need for combined treatment (radiation therapy and radiosurgery), and hormone replacement therapy.
Are there any peculiarities in the diagnosis of craniopharyngeomas?
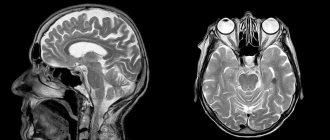
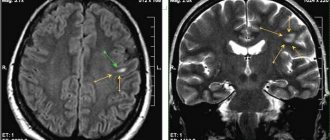
The method of choice in diagnosing CF is an endocrinological examination in combination with MRI of the brain without and with contrast enhancement. A frequent phenomenon on SCT of the brain without contrast and even on plain craniograms is the presence of calcifications in the chiasmatic-sellar region - in children in 85% of cases, in adults in 40%.
What treatment methods are used for craniopharyngiomas?
Surgical removal is the method of first choice in the treatment of newly diagnosed CF, because These tumors, as a rule, are diagnosed already in the presence of a detailed clinical picture against the background of a large tumor size. When removing giant craniopharyngiomas, mortality can reach 5-10%, mainly due to damage to the hypothalamic structures. Small craniopharyngiomas are amenable to surgical removal via a transnasal transsphenoidal approach using neuroendoscopic techniques. Most relapses occur within 1 year, with fewer occurring >3 years (very late relapses occur in cases where “total” excision was intended). With repeated operations, the number of complications and mortality are higher.
Stereotactic radiosurgery is the treatment of choice for the treatment of small recurrent craniopharyngiomas in both children and adults. CF tumors are quite radiosensitive and do not require high doses of radiation. In most cases, even close proximity of the visual pathways does not limit the possibility of effective treatment of the tumor.
Stereotactic radiotherapy is indicated for large CF recurrences (more than 3 cm), persistent recurrence after surgical treatment and/or impossibility of surgical removal. It is carried out in hypofractionation or standard fractionation modes.
Conducting conventional radiation therapy is considered unjustified due to the high risk of endocrine disorders, optic neuritis, and dementia.
In all cases of stereotactic irradiation, cystic craniopharyngiomas can significantly increase in size due to post-radiation changes (not to be confused with continued growth!), which, under conditions of functional saturation of this anatomical area, may require puncture or transnasal emptying of the tumor cyst. This effect is not typical for solid tumors.
Diagnostics
The simplest and most accessible method is skull radiography. The presence of petrification in the chiasmal-sellar region, changes in the shape and size of the sella turcica serve as reliable criteria, which, together with clinical manifestations, make it possible to make a diagnosis.
Changes that are detected using MRI or CT are quite characteristic and in the vast majority of cases allow us to clarify the diagnosis. Each method has its own advantages; CT provides more complete information about the structure of the tumor, the density of the cysts and the presence of areas of calcification. MRI differentiates petrification worse, but tumor measurements and its topography are more clearly revealed.
Treatment
The decision on the nature of the operation and access to the tumor should be strictly individual and determined by the topography of craniopharygiomas and its structure. There are total, subtotal and partial tumor removal. The radicality of the removal is determined based on the surgeon’s impression and data from CT and MRI studies.
Removal of the tumor should be considered total when the control examination does not reveal any remnants and the surgeon is confident that the tumor has been completely removed.
With subtotal removal, the surgeon is forced to leave tumor fragments (capsules) near the bottom of the third ventricle, on large arteries and other important structures.
The surgical approach and removal technique are largely determined by the location of the tumor, so it is advisable to consider them depending on the topography of craniopharyngiomas.
Forecast
If the tumor is removed in a timely manner and there is no malignancy, the prognosis is favorable and return to work is possible.
See also Pituitary gland, tumors; Brain, tumors.
Bibliography:
Arendt A. A. Clinic and diagnosis of craniopharyngiomas, Vopr, neurokhir., t. 21, no. 5, p. 18, 1957; aka, Surgical treatment of craniopharyngiomas, ibid., vol. 22, no. 5, p. 11, 1958; same, Analysis of surgical treatment of craniopharyngiomas, ibid., vol. 23, no. 1, p. 17, 1959; Babchin I.S. On the surgical treatment of craniopharyngiomas, ibid., vol. 16, no. 5, p. 14, 1952; Grekhov V.V. Structure of the parenchyma of craniopharyngiomas (Erdheim tumors), ibid., v. 23, no. 5, p. 1, 1959, bibliogr.; Brain tumors, ed. A. I. Arutyunova, p. 114, M., 1975, bibliogr.; Popov N. A. Tumors of the pituitary gland and pituitary region, p. 106, L., 1956; Bartlett JR Craniopharyngiomas, Brain, v. 94, p. 725, 1971; Clinical microneurosurgery, ed. by W. Th. Koos ao, Stuttgart, 1976, bibliogr.; Koos W. Th. a. Miller M. H. Intracranial tumors of infants and children, p. 188, Stuttgart, 1971; Matson DD a. Сrigler JF Management of craniopharyngioma in childhood, J. Neurosurg., v. 30, p. 377, 1969; Rand RW Micro-neurosurgery, St Louis, 1969; Wertheimer P. et Corradi M. Les craniopharyngeomes aprfcs 40 ans, Neurochirurgie, t. 3, p. 3, 1957, bibliogr.; Ziilch KJ Kraniopharyngeome, Handb, d. Neurochir., hrsg. v. H. Olivecrona u. W. Tonnis, Bd 3 S. 504, B. ua, 1956.
A. A. Arendt, A. H. Konovalov.