Reasons for development
Retinal pigment degeneration is a disease that results in a gradual narrowing of the visual fields. One of the obvious symptoms of the disease is loss of vision during twilight hours. The disease may be caused by a malfunction of a specific gene. In rare cases, the interaction of several genomes is disrupted. The disease is hereditary and transmitted through the male line. The disease may be accompanied by disruption of the hearing aid.
The causes of malfunctions in the functioning of the gene system of the human body have not yet been identified. Overseas researchers have found that DNA abnormalities are not 100% responsible for the development of pigmentary dystrophy. According to experts, the disease provokes disturbances in the vascular system of the eyeball.
Despite the fact that the causes of the disease remain a medical mystery, experts have fairly reliably studied the issue of its development.
Retinal pigmentary degeneration is a fairly rare disease that leads to poor vision in the dark.
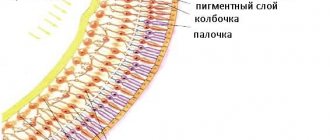
At the initial stage of the disease, a process of metabolic failure occurs in the retina of the eyeball. Disorders also affect the vascular system. As a result of the development of the disease, the layer of the retina in which the pigment is located begins to deteriorate. Sensitive photoreceptors, rods and cones, are located in the same layer. At the first stages, degeneration processes affect only peripheral areas of the retina. That is why the patient does not experience discomfort or pain. Gradually, the changed area begins to increase in size until it covers the entire area of the retina. When the retina is completely affected, the first serious symptoms of the disease begin to appear, a deterioration in the perception of colors and their shades.
The disease can spread to only one eye, but there are often cases when the disease affects two visual organs at once. The first symptoms of the disease appear in early childhood, and by the age of twenty a person may lose his ability to work. Severe stages of retinal pigmentary dystrophy may be accompanied by complications such as cataracts and glaucoma.
Symptoms
The sluggish development of the disease leads to the fact that most patients seek the help of specialists when pathological changes have begun their rapid development. The first serious symptom of the disease is difficulty navigating in low light conditions. Pathologies occurring in the peripheral part of the retina lead to narrowing of the visual fields.
Given the nature of the disease, the main group of patients are children under school age. At this age, minor vision problems are not noticed, which means that parents may not be aware of the development of the disease.
The first stages of development can take a long time - up to five years. Subsequently, degeneration of the peripheral region of the retina begins to progress. The visual fields at this point are greatly narrowed; some patients experience a complete absence of lateral vision. An examination by an ophthalmologist can reveal areas with pathological changes, but if left untreated, they will soon spread throughout the entire retina. At this stage, gaps may appear in some parts of the retina. The vitreous body begins to lose its transparency, becoming cloudy yellow. At this stage, central vision is not affected.
The exact cause of the disease has not been established, but ophthalmologists give only versions of the development of retinal pigmentary degeneration
The disease in an advanced stage can be complicated by the occurrence of diseases such as glaucoma and cataracts. With complications, central vision very sharply loses its sharpness, and over time it can be irretrievably lost. Complications lead to the development of vitreous atrophy.
There is another form of retinal degeneration - atypical. As a result of the disease, the appearance and structure of the vascular system changes. The patient experiences difficulty in orienting in low light conditions.
One of the rarest types of retinal degeneration is a unilateral form, and the patient necessarily develops cataracts.
Treatment of pigmentary dystrophy
Treatment of retinal pigment degeneration, which is in the developmental stage, is most often carried out with the help of medications. The actions of the drugs should be aimed at normalizing blood circulation and nutrient metabolism in the retina and vascular system. In most cases, specialists prescribe the following drugs:
- "Emoxipin". This drug corrects microcirculation in the body.
- "Taufon" . Eye drops stimulate regeneration processes in eye tissues.
- "Retinalamine." The drug, prescribed for retinal dystrophy, has a regenerative effect.
- A nicotinic acid. A vitamin that stimulates the metabolism of nutrients in the body and blood circulation.
- No-shpa with papaverine. An antispasmodic that relieves pressure in the vascular system.
These drugs can be prescribed by a doctor either in the form of tablets, injections or eye drops.
As the disease develops, loss of peripheral vision is determined.
Very often, in addition to medication treatment, a course of physiotherapy is prescribed to stimulate the processes of restoration and regeneration of the retina. Completing this course can activate the functioning of photoreceptors. Some of the popular techniques today are stimulation with electrical impulses, magnetic resonance and ozone therapy. If the disease has damaged the choroid of the eye, it makes sense to undergo surgical intervention.
With the help of surgery, specialists normalize blood circulation in the retinal layer of the eyeball. To achieve this goal, it may be necessary to transplant certain tissues of the eyeball under the perichoroidal space.
Retinal disinsertion.
Causes.
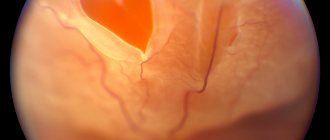
This disease can occur with eye injuries, high myopia, changes in the vitreous body and choroid (adhesions, hemorrhages, tumors). The basis of retinal detachment is the formation of its smallest breaks. These ruptures appear in patients after an eye injury, and in people with high myopia simply when coughing, physical activity, or lifting heavy objects. Through the tear, aqueous humor enters under the retina. Since the retina is loosely adjacent to the choroid throughout its entire length (attached only at the dentate line and the optic nerve), aqueous humor, once under the retina, peels it off in the form of a bubble of various sizes and shapes.
Clinic.
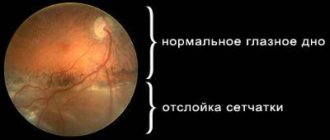
A patient with retinal detachment complains of decreased vision, distortion of objects, loss of vision (a “curtain” appears on one side). Retinal detachment can be suspected if these complaints appear in a patient after a fall, eye injury, or physical activity, especially in people suffering from myopia. A very characteristic symptom of retinal detachment is a narrowing of the visual field. In order to identify a narrowing of the visual field during retinal detachment, sometimes it is enough to carry out an indicative study, comparing your field of vision with the patient’s field of vision.
The diagnosis is made by an ophthalmologist after examining the fundus and ultrasound examination of the eye.
Emergency first aid consists of transporting the patient to the eye hospital as soon as possible under conditions of maximum gentleness, so as not to cause an increase in detachment due to shaking or sudden movements. For transportation, a binocular bandage is applied.
Treatment of retinal detachment is surgical.
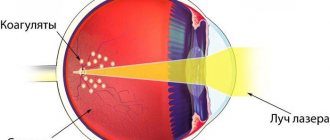
- carry out diathermo-, laser- or photocoagulation,
- according to indications, one of the types of surgical interventions is performed (shortening the sclera, non-through resection of the sclera with filling with catgut, cartilage, circular indentation along the entire circumference in the equator area of the eye, etc.).
Care for patients with retinal detachment is carried out in an eye hospital. Before surgery, patients are prescribed strict bed rest for 5-7 days in order to create better conditions for the retina to fit, determine the boundaries of the detachment and select the surgical method. During this period, patients need good and systematic care (toilet, food, etc.). On the eve of the operation, they are given a cleansing enema and food is limited. After the operation, strict bed rest is prescribed for 10-12 days, sometimes with a forced position of the head - tilting it in a certain direction, depending on the location of the rupture.
All movements are prohibited. After the operation, the doctor and nurse give the patient’s head the desired position using special pillows. The nurse should ensure that the patient does not change this position without the doctor's permission. Due to prolonged forced bed rest, it is necessary to especially carefully monitor the condition of the skin and mucous membranes of such patients, wipe the skin with camphor alcohol, and avoid folds and crumbs on the sheets. You can feed the patient only in a supine position; you need to monitor your stool and urination.
After 2 weeks, the patient is transferred to semi-bed rest, then to the general regime. Sometimes he is prescribed special (hole-shaped) glasses. The nurse should ensure that the patient does not remove these glasses, bend over, or lift heavy objects.
Prevention of retinal detachment consists of timely identification and treatment of eye diseases that can lead to detachment, and in patients with high myopia, adherence to a regimen of physical activity with limited lifting of heavy objects, jumping, and sudden tilting of the head.
The use of vision correcting devices
Some experts recommend treating retinal pigmentary dystrophy using photostimulation devices. Their work is based on a technique that causes stimulation in certain areas of the eyeball and slows down the development of the disease.
The radiation emitted by the equipment stimulates blood circulation in the vascular system of the eyeball and also normalizes nutrient metabolism. Using this technique you can also remove swelling from the retina of the eyeball. Photostimulation of the retina of the visual organs can have a beneficial effect on strengthening the retina and improving the circulation of nutrients in the inner layers of the eyeball.
Damage begins in the periphery and spreads over several decades to the central zone of the retina
How are diseases of the pigment layer diagnosed?
All diseases of this layer of the retina are diagnosed only after a thorough ophthalmological examination. In young children, making an accurate diagnosis can be quite difficult. If you notice that the child has difficulty oriented in the twilight or at night, he should be shown to a doctor: he is probably developing the initial stage of retinal pigment layer degeneration.
Diagnosis of diseases of this element of the visual organs is carried out using the following methods:
- study of visual acuity (both normal and peripheral);
- examination of the fundus of the eye;
- electrophysiological examination;
- study of the degree of adaptation of the eye to darkness.
Forecast
Unfortunately, today medicine is still quite far from resolving the issue of when a disease is in an advanced state. Very often there is news that foreign researchers have found a way to restore certain genes responsible for the occurrence of the disease. Already today, special implants capable of replacing the retina are undergoing the final stage of testing.
Another approach by specialists has revealed that it is possible to completely restore lost vision with the help of injections of a special substance containing cells sensitive to light. However, this technique is still at the experimental stage, and it is still unknown whether scientists will be able to achieve the required result.
Many of those who have encountered this disease know that the prognosis for successful treatment in most cases is unfavorable.
But if the disease is detected at an early stage, using certain treatment methods, its progression can be stopped. In some cases, specialists achieved truly tangible results. People diagnosed with the disease must avoid prolonged physical activity, as well as stress on the visual organs. Facebook
Anomalies of retinal development (coloboma, dysplasia, hemangioma)
Anomalies of the eye membranes are detected immediately after the birth of the child.
They can occur due to gene mutations, chromosomal abnormalities, and toxic effects in the prenatal period of exogenous or endogenous factors. In addition, infectious diseases that the mother suffers from during pregnancy are of great importance. Also influenced by negative environmental factors affecting the embryo, toxins, medications, radiation, etc.
Particularly severe changes in the ocular environment are detected when the fetus is exposed to harmful factors in the early stages of pregnancy. As a rule, the following infections have a negative impact: rubella, syphilis, toxoplasmosis, cytomegalovirus, herpes, HIV.
Among the drugs and substances, the most dangerous include: cocaine, thalidomide, ethanol.
Retinal abnormalities
Congenital anomalies of the retina include: coloboma aplasia, hypoplasia, dysplasia, albinism. In addition, congenital defects are: hyperplasia of the pigment epithelium, vascular anomalies, myelinated nerve fibers, phakomatoses.
Retinal coloboma is the absence of retinal tissue in a limited area. As a rule, it is accompanied by colobomas of the iris and choroid; it can be localized in the center of the eye or on the periphery of its lower half. The cause of coloboma is incomplete closure of the embryonic cleft.
With ophthalmoscopy, this anomaly appears as a white, limited oval or round area near the optic nerve head, sometimes adjacent to it. In the absence of the retina and choroid, the sclera is exposed.
Coloboma is often accompanied by microphthalmos, defects in skeletal development, etc.
Retinal dysplasia is an anomaly that occurs during fetal development as a violation of the ratio of cell elements. Dysplasia may be manifested by non-adhesion of the retina.
This is a rare pathology that occurs due to insufficient invagination of the optic vesicle and is one of the signs of trisomy 13, as well as Walker-Warburg syndrome.
It is detected in combination with other defects of the eye, muscle tissue, and cerebellum.
Albinism is a genetically determined developmental disorder of the visual system that occurs due to impaired melanin synthesis.
People with albinism may also suffer from nystagmus, refractive errors with astigmatism, weak fundus pigmentation, central retinal dysplasia, and changes in the optic chiasm.
This anomaly is also characterized by defects in color vision, changes in brightness and contrast, interhemispheric asymmetry of VEP and supernormal ERG.
Albinism can be tyrosine-negative or tyrosine-positive. In the first case, the occurrence of the disease is associated with the lack of production of the enzyme tyrosinase, as well as the pigment melanin. People with this disease have white skin and hair and do not tan.
Their light iris is easily visible, the reflex from the fundus of the eye is colored bright pink and is visible from a distance. In the tyrosinase-positive form of albinism, on the contrary, the ability to produce melanin is preserved, but its normal accumulation is impossible. The skin of such patients is capable of tanning, but is poorly pigmented.
The hair has a very light or yellowish tint, visual impairment is less severe.
To date, albinism has no cure. Medical care consists of spectacle correction of refractive errors with optical glasses with light filters against the damaging effects of bright light on the retina.
Congenital hyperplasia of the pigment epithelium is characterized by focal hyperpigmentation of the retina. The grouping of pigment spots has the shape of a bear's track with single and multiple foci. There are no changes in the retina around them. Enlargement of foci of pigmentation occurs rarely; they are practically not susceptible to malignancy.
Developmental anomalies also include myelinated nerve fibers. They can be classified as defects in the development of the retina or defects in the development of the optic nerve.
The myelin covering of the optic nerve fibers should normally end at the far edge of the lamina cribrosa. In some cases, it continues up to the nerve fibers of the second-order retinal neurons, bypassing the optic disc.
When performing ophthalmoscopy, the fibers appear as white, radially located shiny stripes that extend to the periphery of the optic nerve head. Sometimes they are not connected to the optic disc.
As a rule, such an anomaly is not accompanied by any symptoms, although in some cases scotomas may occur.
Anomalies of the retinal vessels of a congenital nature manifest themselves as grape-shaped angioma, capillary hemangioma of Hippel-Lindau, Coats disease, as well as cavernous hemangioma, retinopathy of prematurity, miliary Leber aneurysms, capillary hemangioma, parafoveal telangiectasia, etc.
Grape-shaped angioma is, as a rule, a unilateral defect, which during ophthalmoscopy has its own characteristic signs: dilated and significantly tortuous arteries and veins, arteriovenous shunts. In the presence of cerebral vascular pathology with decreased central vision, they speak of “Waburn-Mazon syndrome.” In most cases, treatment is not carried out, since the disease does not progress.
Coats disease is a congenital vascular anomaly with characteristic retinal telangiectasia, aneurysms of various sizes with exudate separation, which over time leads to retinal detachment. Another name for the disease is “external hemorrhagic retinitis.” As a rule, it has a one-sided course and is detected in children (usually boys) at an early age.
Ophthalmoscopy reveals the deposition of a hard, bright yellow exudate in the subretinal space of the posterior ocular pole. In the later stages of the disease, cataracts, glaucoma occur, and subatrophy of the eye occurs. With moderate severity of the pathology, only telagyectasia is observed.
When diagnosing, such an anomaly must be differentiated from tumor processes hidden by exudate or a detached retina, as well as retinopathy of prematurity.
Treatment is the elimination of exudation through obliteration of abnormal vessels using laser coagulation or cryopexy. If large-scale exudative retinal detachment is observed, surgical treatment is prescribed.
Phakomatoses are congenital malformations with ophthalmological and systemic manifestations. The most common among them are: hemangiomas, humartomas, and nodes.
Phakomatoses include Recklinghausen neurofibromatosis, Hippel-Lindau disease, and tuberous sclerosis, which occur according to an autosomal dominant principle. In addition, they include Sturge–Weber–Krabbe syndrome.
The cause of the disease is a mutation of the gene identified in all cases of dominant diseases.
Recklinghausen's neurofibromatosis is characterized by a tumor of Schwann cells, with a skin manifestation - the appearance of multiple fibromas. The gene responsible for the development of the disease is located on chromosome 17. The disease manifests itself as deforming neuromatous elephantiasis, which is provoked by neurofibromatous infiltration.
When diagnosing, pay attention to café-au-lait spots on the skin. There must be at least 6 of them, over 1.5 cm in size.
Among the ophthalmological manifestations of neurofibromatoses, the following are noted: plexiform neurofibroma of the orbit and eyelids, congenital glaucoma, S-shaped palpebral fissure, iris hamartomas and hamartoma infiltration into the choroid, conjunctival neurofibroma, prominence and thickening of the corneal nerves, optic nerve glioma, buphthalmos, pulsating exophthalmos.
Hamartoma is a tumor of embryonic tissue with delayed differentiation relative to the differentiation of the organ on which it develops. The cells of such a tumor do not have abnormalities in the structure, but there have been changes in the density of cell populations.
Melanocytic hamartomas, called Pisch's nodules, are detected on the iris of adult patients even before skin manifestations of the disease and are a diagnostic sign.
Plexiform neurofibroma is a tangle of hypertrophied, intertwined nerves. Due to the proliferation of Schwann cells, they appear lumpy.
A particularly common complication of neurofibromatosis type 1 is vascular disorders.
These include: occlusion of blood vessels or narrowing of their lumen, which subsequently lead to perivascular fibroglial proliferation.
Some of the signs of oxygen starvation of the retina are considered to be avascular zones along its periphery, preretinal fibroglial membranes, arteriovenous shunts, and optic nerve atrophy.
If the tumor causes deformation of adjacent tissues or dysfunction of an organ, it must be removed.
Neurofibromatosis type 2 is a rare disease. Its typical symptom is schwantomy (usually bilateral) of the eighth pair of cranial (auditory) nerves. Ophthalmological complications include combined hamartomas of the retinal pigment epithelium, optic nerve meningioma, and glioma.
Hippel-Lindau disease is a genetically determined disease with a defect in chromosome 3p25. As a rule, its discovery occurs by chance when a child undergoes a diagnostic examination for strabismus.
Retinal angiomas are characterized by the shape of cherries, which are equipped with developed vessels that feed and drain it. Similar formations are retinal hemangioblastomas, since they are histologically similar to the latter that form in the cerebellum.
Retinal hemangioblastomas are characterized by exophytic and endophytic growth involving the disc and the optic nerve itself. Their combination with maculopathies is often detected.
Retinal angiomatosis can also accompany renal cystosis, renal carcinoma, pheochromocytoma, etc.
Due to increased vascular permeability, retinal exudate with a large amount of lipids can accumulate inside, which leads to the development of exudative retinal detachment in the last stages of the disease.
When diagnosing arteriovenous FA, the concentration of contrast agent in the angioma is detected. In the later stages of the disease, fluorescein goes beyond the vessels into the surrounding tissues, which is due to the defectiveness of tumor vessels.
Treatment options include laser coagulation, cryotherapy, and surgical tumor removal.
Tuberous sclerosis, also known as Bourney-Ville disease, is a rare autosomal dominant disease that occurs due to the malfunction of two genes on chromosomes 9 and 16.
The classic triad of pathology is epilepsy, facial angiofibromas, mental retardation. The retina is affected by whitish tumor formations localized near the optic nerve head, vaguely reminiscent of mulberries. Astrocytomas, which are called giant drusen, can be detected on the optic nerve head. They are sometimes mistakenly associated with retinoblastoma.
The treatment option is to place patients in a neurological clinic. The prognosis of the disease is serious: with an increase in neurological manifestations, death soon occurs.
Source: https://retina-center.ru/articles/102-anomalii-razvitiya-glaza